La leyenda de la Eva africana
Vivo en California desde hace 25 años, pero todavía tengo que hacer un esfuerzo para recordar que aquí la enseñanza de la teoría de la evolución no es libre. La informática avanza a gran velocidad, se construyen los aviones más sofisticados del mundo, pero un gran sector de la población todavía aprende la historia de la humanidad a partir de una interpretación literal de la Biblia. Los movimientos religiosos fundamentalistas son muy fuertes y pretenden abolir la enseñanza del evolucionismo; como no pueden, tratan de ponerle trabas y piden que, como mínimo, se presente también su verdad, la creación, y se le dedique la misma cantidad de tiempo. Para muchos estadounidenses el mundo fue creado el año 4004 antes de Cristo. Para esas mismas personas nuestro trabajo, evidentemente, es absurdo y probablemente blasfemo.
La fe religiosa puede hacer que el hombre afronte cualquier sacrificio, incluyendo el de su vida y sus propias ideas. La probabilidad de convertir a un creyente fundamentalista a la teoría de la evolución me parece demasiado pequeña como para que valga la pena dedicarle algún esfuerzo, por lo menos aquí. Por lo tanto, partiremos de la base de que ha habido evolución y de que todas las personas sin prejuicios aceptan esta idea sin problemas. Hay que decir que, aunque fuera verdad que el mundo fue creado el 4004 a.C., de todos modos ya no seríamos los mismos de entonces. En 6000 años no ha podido haber mucha evolución, ya que es lenta; pero inexorable.
En este capítulo veremos la extensión de los estudios con marcadores (sinónimo de polimorfismos) más potentes de los que hemos visto hasta ahora, pero sobre los que poseemos un número menor de datos. De todos modos, con los que hay podemos llegar a conclusiones importantes y estudiar el comportamiento de varios modos de reconstrucción de los árboles evolutivos. Estas diferencias nos proporcionan otras informaciones importantes.
El estudio de un gen a partir de su ADN, o de la proteína que produce el gen, revela que en las especies actuales se han acumulado mutaciones que se pueden contar fácilmente. El análisis de la diferencia de mutaciones acumuladas entre dos especies permite calcular el tiempo evolutivo que las separa; este modo de reconstruir los tiempos de evolución se denomina reloj molecular. Nos ha revelado que hay que volver atrás por lo menos 5 millones de años para encontrar un antepasado común del hombre y de nuestro primo más cercano, el chimpancé. Hay que ir aún más atrás para encontrar una rama evolutiva que nos lleva a un primo un poco más lejano, el gorila, y aún más, 14 millones de años, para llegar al más lejano, el orangután, cuyo parecido con nuestra especie sigue siendo sorprendente, a pesar de su largo pelaje rojo. Este «pariente» nuestro vive en el sureste de Asia, mientras que los más cercanos son africanos. Este hecho convenció a Darwin de que el hombre se había originado en África. Ahora sabemos que los australopitecos, descendientes del progenitor común del hombre y el chimpancé, entre los que hay antepasados directos del primero, también vivían en África. El primer antepasado que se considera digno de pertenecer al género Homo tiene más o menos 2,5 millones de años (H. habilis). Fabricaba instrumentos de piedra muy primitivos, era completamente bípedo, tenía la cabeza más grande que sus inmediatos predecesores o los grandes simios actuales, pero aún muy pequeña comparada con la nuestra. No hay discrepancias significativas sobre el hecho de que se desarrollara en África, donde le siguió Homo erectus. Este último fue el primer antepasado que empezó a explorar y a ocupar todo el Antiguo Mundo. Se creía que esta expansión se produjo hace 1 millón de años, pero los descubrimientos recientes tienden a retrasar esta fecha a unos 2 millones de años atrás. El cráneo de Homo erectus es más grande que el de sus predecesores, pero el volumen semejante al nuestro sólo se alcanza con la última especie, Homo sapiens. Al principio, los rasgos de Homo sapiens aún eran un poco simiescos, y sólo hace 100 000 años aparecieron cráneos que se podrían confundir con los actuales. El primer hombre anatómicamente moderno ha sido descrito en el sur y el este de África, donde la geología volcánica ha favorecido casi todos los hallazgos paleoantropológicos importantes.
El descubrimiento de cráneos de hombre moderno, más o menos de la misma edad, en Oriente Próximo, ha causado cierta confusión. Es verdad que Oriente Próximo está bastante cerca de África y se comunica directamente con ella, sin tener que atravesar el mar, por lo que casi se trata del mismo país. Pero inevitablemente surgen dudas sobre el origen preciso del hombre moderno. ¿Oriente Próximo o África? Tampoco podemos decir, admitiendo que la respuesta correcta sea África, si se trata de la oriental o la meridional, ya que se han encontrado hombres modernos de hace 100 000 años en las dos regiones. En realidad, la historia es más complicada: en Oriente Próximo se han encontrado hombres de Neandertal de hace 60 000 años, y parece que en esa época los hombres modernos habían desaparecido. Richard Klein cree que la explicación de este fenómeno es que la primera colonización del hombre moderno fuera de África, a Oriente Próximo, hace 100 000 años, no tuvo éxito. Este fracaso habría facilitado la colonización de Oriente Próximo, hace 60 000 años, por el hombre de Neandertal (que primero vivía en Europa y luego se había desplazado hacia el este y el sureste). Aunque en esta región no hay rastro de hombres modernos en dicha época, los hallazgos que permiten una buena datación son tan escasos que la falta de restos de un tipo humano determinado no demuestra su inexistencia. Sea como fuere, más tarde el Neandertal se retira y el hombre moderno ocupa toda el área.
Debió de haber buenos motivos para esta expansión. Seguramente entre los más importantes estaban las novedades tecnológicas que enriquecieron la alimentación. Observaciones recientes realizadas en África muestran que el hombre moderno de hace 100 000 años ya tenía una tecnología bastante avanzada para conseguir comida. Pero también pudieron influir la posibilidad de desplazarse, de sobrevivir en condiciones climáticas nuevas, y muchas otras conquistas nuevas.
El cerebro humano aumentó continuamente hasta Homo sapiens, hace unos 300 000 años, cuando su crecimiento, a juzgar por las dimensiones del cráneo, se detuvo. Como en el caso de los ordenadores, el hardware creció, pero con eso no bastaba, hacía falta que el software se modificara, se hiciera más complicado y potente. Conocernos bien la diferencia entre nosotros y nuestros vecinos más cercanos en la evolución, los grandes simios. La innovación importante que distingue al hombre de estos primos lejanos fue la comunicación, posibilitada por un lenguaje, bastante rico y refinado. Los chimpancés y los gorilas pueden aprender a utilizar bastante bien 300 ó 400 palabras, pero los investigadores tienen que recurrir a argucias, como el lenguaje de los sordomudos, o unos ordenadores especiales, porque nuestros primos lejanos no saben usar la lengua, la faringe y la laringe para emitir sonidos comparables a los nuestros, es decir, no tienen el don de la palabra. Pero saben utilizar símbolos para indicar las cosas, y reconocer los símbolos cuando se les habla en una de las lenguas artificiales creadas por los investigadores que han realizado estos interesantes experimentos de comunicación con los animales. Hoy sabemos que, de todos modos, les cuesta mucho componer frases (si no son muy simples), así como entender la gramática y la sintaxis. En cambio, todos los hombres modernos que viven actualmente usan lenguas complejas, con varios miles de vocablos, con una gramática y una sintaxis difíciles, tan avanzadas y capaces de expresar conceptos especiales como las lenguas más difundidas a las que estamos acostumbrados. No existen lenguas «primitivas»; las 5000 que hoy existen en la Tierra son igual de ricas, por lo menos en potencia. Cualquier persona dotada de una inteligencia normal (es decir, que no tenga deficiencias mentales, como las que aparecen en una proporción muy pequeña de individuos de cada población) puede aprender perfectamente cualquier lengua, si lo hace a una edad lo bastante temprana. Pero los niños que han pasado sus primeros cuatro, cinco o seis años sin aprender ninguna, por falta de enseñanza, ya no pueden alcanzar el conocimiento perfecto de una lengua, cualquiera que sea. Si la enseñanza empieza aún más tarde pueden ser incapaces de articular palabra. Todo esto no tiene nada que ver con el estudio de un segundo idioma, que se puede aprender a cualquier edad, después de dominar la lengua materna. Sólo hay una limitación en el aprendizaje de una segunda lengua: después de la pubertad, a casi todos les resulta muy difícil aprender a pronunciar los sonidos de la lengua extranjera que no existen en la suya. He aquí una buena razón para introducir la enseñanza de lenguas extranjeras en primaria y secundaria. Por desgracia, la mayoría de los gobiernos hacen caso omiso de esta regla casi absoluta.
Otras razones han hecho pensar que la lengua del hombre moderno se desarrolló, hasta alcanzar el nivel actual, en el período comprendido entre 150 000 y 70 000-100 000 años atrás. El arqueólogo Glynn Isaac descubrió que las culturas paleolíticas africanas de esta época presentan una alta diferenciación local. Lo demuestra de un modo indirecto el número de nombres distintos que los arqueólogos han tenido que dar a las civilizaciones africanas de este período. Isaac establece un paralelismo entre el incremento de la variación local de las culturas líticas y la diferenciación local de las lenguas y los dialectos que siguió al aumento de complejidad de las lenguas. La posibilidad de comunicarse de un modo más refinado, gracias a lenguas casi tan perfeccionadas como las actuales, debió ser de gran ayuda en los viajes de exploración y colonización de este antepasado tan cercano a nosotros, el hombre anatómicamente moderno. Hace 60 000-70 000 años empezó a viajar partiendo de África, hasta alcanzar en relativamente poco tiempo los lugares habitables más alejados del globo, como Tierra del Fuego, la costa del océano Ártico, Tasmania y, por último, Groenlandia.
Una lengua más perfeccionada (hipotética, para ser rigurosos) no fue lo único que ayudó a los primeros hombres modernos en esta empresa extraordinaria. En los últimos 100 000 años empezó una mejora de las técnicas de trabajo de los utensilios de piedra que produjo un cambio de estilo de los instrumentos líticos, una difusión mayor de instrumentos hechos con otros materiales (madera, marfil) que tenían muchas ventajas, pero por desgracia resisten peor el paso del tiempo y se han conservado sólo ocasionalmente: es el paso de la técnica llamada «musteriense» a la «auriñaciense». Pero aún más importante tuvo que ser la navegación. No nos han llegado restos de embarcaciones o balsas que, al ser de madera, han tenido pocas posibilidades de conservarse hasta nuestros días, pero sabemos que para pasar del sureste de Asia a Australia el hombre moderno tuvo que atravesar cinco o seis trechos de mar de unos 60 kilómetros de longitud (no es mucho comparado con lo que se consigue hacer hoy, pero entonces requería técnicas nuevas).
Desgraciadamente, las principales fechas referentes al hombre moderno son más antiguas que las que podemos establecer por medio del radiocarbono, cuyo límite (como ya se ha dicho) es de 40 000 años. Pero hoy existe un perfeccionamiento del radiocarbono, que permite llegar hasta 60 000 años, y hay métodos nuevos (la termoluminiscencia y otros afines) que permiten ir aún más atrás. Pero acabamos de empezar a utilizarlos, y aún no conocemos sus limitaciones.
La arqueología nos da algunas ideas sobre las fechas de ocupación de los distintos continentes, que se pueden confrontar con las distancias genéticas entre ellos. Cuanto más antigua es la fecha de entrada, más tiempo hubo para que surgieran diferencias genéticas entre los colonizadores de un continente y sus habitantes. Cabe esperar, pues, que la diferencia medida de la distancia genética entre continentes en tiempos más lejanos sea mayor que la que existe entre continentes ocupados más tarde. Cuando el hombre moderno salió de África, probablemente entró primero en Asia. Hace 100 000 años es la fecha aproximada para la ocupación de Oriente Próximo. Pero ¿cómo llegó a la parte oriental de Asia? Pudo recorrer la costa de Arabia, y luego la de la India, hasta el sureste asiático. Desde allí el camino se ramifica: hacia el sur, a Nueva Guinea y Australia, y hacia el norte, a China, Japón y por último Beringia y América. Hay dos argumentos a favor de esta ruta costera: en la última parte, del sureste asiático a Australia, el hombre moderno sin duda era ya capaz de atravesar largos trechos de mar, y no es descabellado pensar que ya poseía esta técnica al salir de África. El clima y la alimentación (a base de marisco, pescado, etc.) habrían sido los mismos durante todo el trayecto, reduciendo los problemas de adaptación a los distintos medios. Una alternativa a la ruta costera habría sido un recorrido al sur o al norte de las cordilleras de Asia central, pero no hay ningún rastro de su paso por una ruta terrestre hace 60 000 o 70 000 años, cuando tuvo que producirse. Sabemos muy poco sobre la llegada a Asia oriental, a excepción de una fecha que sitúa al hombre moderno en China hace 67 000 años. En Australia hay ejemplares fósiles de hombre moderno de hace 35 000-37 000 años, pero otras fechas, calculadas más recientemente con la termoluminiscencia en objetos de fabricación humana, señalan su llegada entre 50 000 y 60 000 años atrás. La separación entre el sureste asiático y Australia se puede remontar, pues, a 60 000 años atrás.
La entrada en Europa, probablemente a través de Asia occidental, es algo anterior a la desaparición del neandertal, y data de unos 40 000 años. La entrada en América, sin duda desde el noreste asiático a través de Alaska, es la más difícil de datar. Los científicos sugieren fechas muy distintas para la primera entrada en América, que van de 15 000 años a 30 000 e incluso 50 000.
Según la hipótesis más sencilla, las distancias genéticas entre dos continentes contiguos deberían ser proporcionales a la antigüedad de la ocupación. En el capítulo anterior hemos visto que dos continentes adyacentes, uno de los cuales es el punto de partida y el otro el punto de llegada, muestran las distancias genéticas del cuadro de la página 45, cuyo promedio se recoge en el cuadro siguiente. Hemos añadido las presuntas fechas de la primera ocupación de los continentes.
| Continentes | Distancia genética | Primera fecha de ocupación (miles de años) | Proporción |
| África-Asia | 206 | 100 | 2,06 |
| Asia-Australia | 101 | 55 | 1,84 |
| Asia-Europa | 97 | 43 | 2,26 |
| Asia-América | 89 | 15-50 | 5,93-1,78 |
Las tres primeras distancias genéticas corresponden más o menos al doble de la fecha de ocupación; la media de las tres proporciones: distancia genética dividida por el tiempo de evolución (2,06; 1,84; 2,26) es 2,05. En lo que se refiere a América, las fechas no son satisfactorias, pero 15 000 años parecen demasiado pocos. De acuerdo con la proporción 2,05 de los primeros tres valores, la fecha de entrada en América se puede calcular en 89/2,05 = 43 000 años. Conviene puntualizar que probablemente la distancia entre los amerindios y los asiáticos que se recoge en el cuadro es demasiado grande, ya que se basa en los análisis de toda Asia, mientras que en la colonización de América sólo participó Asia oriental. Sería mejor tomar en consideración la distancia entre los asiáticos del este y los amerindios, más pequeña (igual a 66). Se obtiene así una fecha de entrada de 66/2,06 = 32 000 años.
Hasta ahora nuestro árbol no plantea problemas: las fechas de ocupación de los continentes están de acuerdo con las distancias genéticas observadas.
Uno de mis principios al empezar este trabajo era que sólo los caracteres determinados totalmente por los genes podían dar respuestas satisfactorias para el estudio de la evolución. Por este motivo no me fiaba de caracteres como la estatura y las distintas medidas antropométricas, dado que están influidas tanto por los genes como por las condiciones de crecimiento, como la alimentación y el clima, y por eso pueden cambiar en un corto espacio de tiempo bajo la acción de estos factores ambientales. Además, el medio también puede modificar su base genética a largo plazo, a través de la selección natural. Pero se entiende fácilmente que la divergencia que puede producirse en la evolución, por ejemplo, entre poblaciones que viven en el hemisferio norte o sur con respecto a las que viven junto a los trópicos, puede complicar el análisis de la historia evolutiva. Los caracteres somáticos sometidos a una fuerte selección debida al medio sólo reflejan las condiciones ambientales que conocieron estas poblaciones en el período más reciente. Pero no nos pueden decir exactamente cuál fue la duración de ese período, ya que en general desconocemos cuánto tiempo tiene que pasar para que cambie un tipo físico sensible a determinadas condiciones ambientales. Por consiguiente, los genes que se prestan mejor al estudio de la evolución son los no sensibles a la selección natural: genes incapaces de funcionar, como los seudogenes, y en general todos los segmentos del ADN desprovistos de actividad biológica (si existen y es posible identificarlos). Están más sometidos a la deriva genética y, en general, al azar, y son más útiles que los sometidos a la selección natural, que en cambio nos hablan del medio vital. Recordemos que se llaman selectivamente neutros. Ya Darwin se había percatado de ello, y decía que los caracteres más útiles para reconstruir la evolución son los más corrientes e insignificantes.
Desde los primeros análisis de los árboles evolutivos, en 1963, nos pareció importante realizar un estudio similar al de los genes, utilizando los caracteres antropométricos clásicos: el color de la piel, los datos de la talla y las principales medidas antropométricas (los llamaremos caracteres morfológicos). Nos basábamos en el principio de que hay que utilizar todos los datos que puedan dar informaciones y que, si alguno de ellos da resultados distintos de los demás, hay que buscar una explicación sólida de las diferencias. Volvimos a utilizar los valores de quince poblaciones, lo más parecidas posible a las que ya habíamos analizado para los genes. No nos sorprendió demasiado que los resultados fueran muy distintos de los obtenidos con los genes. Por ejemplo, los africanos y los australianos aborígenes mostraban un parecido considerable en sus caracteres morfológicos y antropométricos, mientras que en el árbol de los genes las dos poblaciones presentan una divergencia máxima.
La discrepancia entre árboles genéticos y antropométricos tenía explicaciones sencillas. Probablemente, si los caracteres antropométricos habían dado resultados muy distintos de los genéticos, era porque estaban muy influidos por la selección natural debida al clima. Es bien sabido que el color de la piel está determinado en gran medida por la intensidad solar y, en general, por el clima. Además, casi todos los caracteres antropométricos están fuertemente relacionados con la estatura que, en todos los animales, responde notoriamente al clima.
Los caracteres antropométricos y morfológicos como el color de la piel reflejan la acción selectiva de los distintos climas a los que estuvieron expuestos los hombres modernos durante su migración por la superficie de la Tierra. Son mucho menos sensibles a los demás aspectos de la historia de la evolución humana, como las migraciones, que les hicieron desplazarse con menos frecuencia en latitud que en longitud. Los genes, en cambio, son testigos más fieles de esta historia de desplazamientos.
Los datos morfológicos que habíamos utilizado (véase HGHG, p. 71) se habían recabado de gran número de trabajos, y por lo tanto estaban sujetos a los inevitables errores de la heterogeneidad de mediciones hechas por muchos autores distintos. Un antropólogo de Harvard, W. Howells, hizo un estudio muy detallado de gran número de cráneos, a partir de una abundante serie de mediciones hechas por él mismo. El análisis de los datos se hizo con métodos estadísticos modernos, y los resultados fueron muy parecidos (HGHG, p. 72) a los de nuestra primera investigación sobre los caracteres antropométricos. Con un segundo análisis de los mismos datos craneométricos, Howells procuró eliminar la influencia de la estatura general y de las dimensiones, centrándose en el estudio de la forma del cráneo. Pero esta última es muy sensible a la proporción entre medida de la cara y de la bóveda craneal, y también depende de la temperatura, pues en las regiones frías hay una notable reducción de las dimensiones de la cara con respecto a la bóveda, lo que produce un cambio importante en la forma de la cabeza. El uso de índices de la forma, más que de las dimensiones generales, no modificó las conclusiones: caracteres muy influidos por la selección natural debida al clima no pueden proporcionar una descripción fiel de la historia evolutiva completa de la especie, ya que muestran un aspecto parcial, la historia de los medios ocupados por los distintos grupos en el último período durante el cual se diferenciaron de los demás.
Los métodos de reconstrucción de los árboles se han multiplicado. En particular se han creado unos cuantos que se pueden llamar de evolución mínima, porque buscan el árbol que permite llegar de un hipotético antepasado a todas las poblaciones que tomamos en consideración en nuestro análisis con la mínima cantidad de evolución. La descripción de las poblaciones humanas cuya evolución estudiamos se realizó al principio en términos de frecuencias génicas, como en los ejemplos de las frecuencias de ABO y de Rh que hemos dado al principio. Si una población tiene una frecuencia Rh del 40 por 100 y otra del 35 por 100, en algún tiempo pasado hubo un antepasado común que tenía otra frecuencia distinta, tal vez intermedia. ¿Cómo se puede calcular? Podemos hacerlo de acuerdo con ciertas hipótesis, como, por ejemplo, que la evolución ha sido la mínima posible. Anthony Edwards encontró una solución elegante, al principio de nuestro trabajo, pero él mismo admitió que el método sólo es preferible por razones formales, ya que no hay ningún motivo para que la evolución tenga que haber sido la mínima posible. Sobre todo si la variación de las frecuencias génicas es, por lo menos en parte, de carácter aleatorio, como sucede bajo los efectos de la deriva genética, pero no tiene sentido pensar que el camino evolutivo haya sido el mínimo posible. Las frecuencias génicas cambian a lo largo del tiempo de un modo que se podría comparar con el caminar de un borracho, o el movimiento de un cuerpo ultramicroscópico por la colisión aleatoria de las moléculas. Los físicos lo llaman movimiento browniano y su descripción matemática se llama «paseo al azar».
Hay otro tipo de datos genéticos con los que la idea de evolución mínima ha tenido mucha fortuna. En vez de las poblaciones, estudiamos individuos aislados. Sabemos que el ADN está formado por cuatro tipos de nucleótidos distintos: A, C, G y T (véase la nota 1 del capítulo 2, p. 210). Supongamos que un segmento de ADN de seis nucleótidos sea:
| TTACGA | en el hombre | (H) |
| TAACGA | en el chimpancé | (C) |
| TAACGT | en el gorila | (G) |
Sabemos que el gorila es el más viejo de los tres, evolutivamente. De modo que el árbol es:
Si comparamos los tres segmentos de ADN vemos que se produjo una mutación de A a T en el segundo nucleótido, después de la aparición del antepasado común de H y C, y antes del origen del hombre. Pero tuvo que haber por lo menos otra, que podría ser una mutación del mismo nucleótido T a A entre el antepasado común de HGC y el común de HC, aunque también podría tratarse de una mutación de A a T en la rama larga que va del antepasado común de los tres al gorila. Y también es posible que en esta rama larga haya habido otras mutaciones: que en el antepasado común de los tres hubiera otro nucleótido, ni A ni T, por ejemplo C, y que éste mutara dos veces, una a T en la rama del gorila y otra a A en la rama de HGC a HC.
El método de evolución mínima sugiere que el número de mutaciones sea mínimo. En este caso, que sean sólo dos, sin que se pueda especificar (si no se hacen más hipótesis) si la mutación del mismo nucleótido se produjo en la rama larga o en la corta. El método se llama de máxima parsimonia, y ha tenido un gran eco entre un grupo de taxonomistas que han llamado cladística a su disciplina. El nombre procede del libro de un evolucionista alemán, W. Hennig, quien destacó acertadamente la importancia de tener en cuenta los caracteres ancestrales para reconstruir la evolución. Pero la veneración por la máxima parsimonia no está justificada. El sentido común apoya sólo hasta cierto punto la idea de reducir al mínimo el número de mutaciones. El motivo a favor es que éstas son escasas, pero siempre son posibles las mutaciones de vuelta. Se ha demostrado que en el recuento de las mutaciones hecho con el método de la máxima parsimonia hay un error por defecto, porque no tiene en cuenta adecuadamente el hecho de que las mutaciones son reversibles. El método de máxima verosimilitud siempre es el mejor para controlar la validez de una hipótesis.
El problema más grave es que puede haber influencias recíprocas importantes entre las distintas ramas, sobre todo si ha habido mezcla de poblaciones, o también, sencillamente, flujo génico de unas a otras (y no recíprocamente; pero por lo general siempre hay cierta influencia recíproca entre dos poblaciones vecinas, que se manifiesta en el intercambio genético entre ellas, aunque casi nunca es por igual en ambas direcciones). A este respecto puede resultar útil un método de evolución mínima, porque puede poner en evidencia tanto las excepciones de la velocidad de evolución constante, como las excepciones de la falta de mezcla. Hoy el método de evolución mínima más fácil de usar con ordenador es el que han desarrollado Saitou y Nei, también llamado «unión de los vecinos» (neighbour joining, o NJ).
Una propiedad importante es que los árboles NJ y, con ellos, todos los de evolución mínima, muestran grandes diferencias de longitud en sus ramas. Se suele interpretar que la longitud de una rama se debe a la velocidad de evolución en esa población y en el intervalo de tiempo al que se refiere la rama; pero puede haber otras razones. En un momento dado se vio que los árboles NJ solían tener una estructura distinta de la de los árboles obtenidos mediante average linkage o máxima verosimilitud. Esta diferencia es importante, y requiere una explicación. Pero empecemos viendo si la diferencia entre los métodos se representa con marcadores distintos de los clásicos que hemos utilizado hasta ahora.
Hasta ahora hemos discutido los resultados obtenidos con los marcadores clásicos, es decir, los polimorfismos obtenidos con métodos inmunológicos y electroforéticos en productos génicos, proteínas, etc. En los años ochenta ya fue posible estudiar directamente el ADN. Los tipos de polimorfismos estudiados en primer lugar recibieron el nombre de RFLP (restriction fragment length polymorphism, polimorfismo de longitud de los fragmentos de restricción). Al principio, cuando eran los únicos conocidos, tuvieron mucha importancia, pero eran difíciles y largos de usar y requerían mucho ADN.
Dado que por primera vez permitían realizar con bastante eficacia la búsqueda de los genes responsables de enfermedades hereditarias, se produjeron por miles, mientras que hasta entonces los polimorfismos clásicos no llegaban a 300 y no había esperanzas de aumentar su número de un modo sencillo y rápido.
Un problema práctico consiste en el hecho de que por lo general el análisis estadístico de los RFLP requiere gran cantidad de ADN. Por este motivo empezamos a utilizar una técnica mediante la cual algunas células de la sangre se reproducen indefinidamente, con lo que se obtienen cantidades de ADN suficientes para cualquier experimento.
En Stanford y Yale, en colaboración con Judy y Ken Kidd, de la Universidad de Yale, empezamos un programa de recogida de cultivos celulares de varias poblaciones representativas de la especie humana (entre 20 y 60 individuos de cada una). Este trabajo, gracias al cual reunimos una colección de treinta poblaciones, fue un proyecto piloto del programa «Diversidad genómica humana», para el que en 1997 esperamos obtener fondos suficientes en Estados Unidos. Se han iniciado programas parecidos en China, y otro, llamado «historia biológica de las poblaciones europeas», ha empezado con la ayuda económica de la Unión Europea.
Las poblaciones que aparecen en la figura 2 derivan casi por completo de la colección Stanford-Yale de cultivos celulares. Las africanas incluyen dos tipos muy distintos de pigmeos (muestras que obtuve en el suroeste de la República Centroafricana, junto a Bagandou, en 1984, y en Zaire, en Mambasa, en el centro de la selva de Ituri, en 1985). Los pigmeos mbuti de Ituri son los más bajos. Los de la República Centroafricana, algo más altos, están muy mezclados, hasta un 75 por 100, con agricultores de origen bantú y sudanés. Las muestras senegalesas proceden de los mandekalu, una población de campesinos del oeste de África, y fueron recogidas por el equipo de André Langaney. Los europeos analizados descienden de una población protestante de California procedente de Europa (Alemania y Gran Bretaña) y de otra de donantes de sangre de Bérgamo, Italia (estudiados por G. B. Ferrara, de Génova). Los japoneses y los chinos (sobre todo del sur de China) viven en San Francisco, California, pero nacieron en Oriente. Una población de Oceanía está formada por melanesios de la isla de Bougainville, y su sangre fue recogida para nosotros por Jonathan Friedlaender, de Filadelfia. Los australianos y los pueblos de Nueva Guinea proceden de numerosos lugares de esta región, y fueron recogidos por el grupo de Allan Wilson de Berkeley. En otros trabajos hemos utilizado poblaciones indígenas de América del Sur, que en el árbol se sitúan exactamente donde se espera encontrarlas de acuerdo con el análisis hecho con los marcadores clásicos.
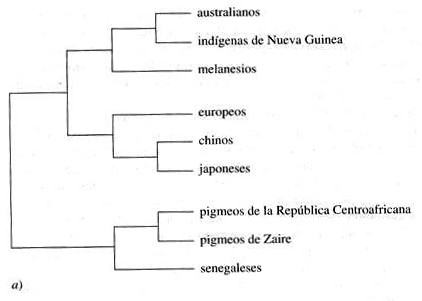
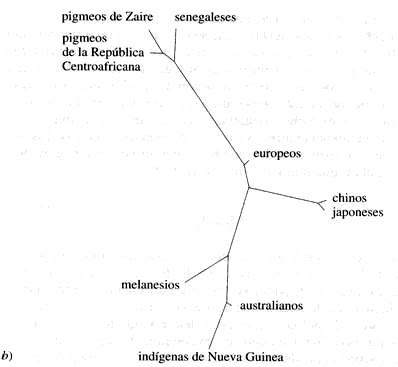
2. Árboles de nuevas poblaciones, obtenidos con marcadores de ADN llamados RFLP. a) Método UPGMA, con velocidad evolutiva constante; b) método NJ, de mínima evolución (según Poloni et al., 1995).
En la figura 2 comparamos dos árboles, obtenidos a partir de 9 poblaciones con 78 polimorfismos de tipo RFLP, recurriendo a dos métodos distintos: el de average linkage o UPGMA (unweighted pair-group method using arithmetic averages, método no pesado de agrupamiento por pares usando medias aritméticas), un equivalente de máxima verosimilitud, y el método NJ. Hay una diferencia importante en los resultados. Los marcadores clásicos también dan resultados similares.
Con los métodos de análisis directo con ADN se descubrieron muchos otros tipos de marcadores. Vamos a describir los datos obtenidos con microsatélites (más adelante explicaremos su naturaleza). Hoy existen muchos miles de microsatélites, casi todos producidos en Francia por una industria genética (la Genethon), con el ADN mitocondrial, lo que ha dado origen a la famosa historia —se podría decir leyenda— de la Eva africana. Ahora conocemos métodos que no existían cuando empezamos a trabajar con los polimorfismos del ADN. Estos polimorfismos, basados en la amplificación del ADN en probeta, gracias a la síntesis catalizada por la polimerasa del ADN (la técnica se llama polymerase chain reaction o PCR), permiten emplear una cantidad mucho menor de ADN.
Ante todo hay que decir que no se encuentran diferencias muy importantes entre las conclusiones obtenidas con los distintos marcadores, si empleamos un número lo bastante grande de ellos. Pero sigue habiendo diferencias en los resultados si usamos métodos distintos para reconstruir los árboles (como hemos visto en la figura 1), aunque se utilicen los mismos marcadores. En realidad, existe una coincidencia casi perfecta entre los resultados obtenidos con los distintos tipos de marcadores, y una diferencia igual de perfecta entre los métodos, que se pueden resumir de la manera siguiente: la primera ramificación en el árbol siempre es la que separa a africanos de no africanos. La segunda es la que separa a Oceanía de otros pueblos (Asia + Europa + América) en el caso de los métodos de velocidad constante, como máxima verosimilitud o average linkage, y la que separa a Europa de los demás (Asia + Oceanía + América) si se usa la mínima evolución. Con NJ, sobre todo, Europa (y en menor medida el este de Asia) tiene una rama muy corta, en algunos casos incluso negativa, un resultado inaceptable.
La coincidencia entre los marcadores y la diferencia de los métodos de reconstrucción de los árboles evolutivos son muy evidentes; está claro que requieren una explicación.
Los factores evolutivos que pueden cambiar mucho de un lugar a otro son dos: la selección natural y la deriva genética. La deriva siempre está presente para todos los genes, y en una población dada tiene la misma intensidad media para todos los genes, ya que es una propiedad de la población, al estar determinada por su dimensión demográfica en la rama evolutiva examinada. La selección natural, por el contrario, puede variar de un gen a otro, en cada población y rama. Pero los genes que presentan una diferenciación elevada, que como hemos dicho pueden estar sometidos a una selección que varía aleatoriamente en el espacio y en el tiempo (como los genes que producen anticuerpos), dan lugar a árboles evolutivos parecidos a los que se obtienen con los otros genes. Es difícil, pues, admitir que una rama sea larga o corta a causa de una diferencia de intensidad de selección.
La posibilidad de que la deriva sea la causa de la variación de longitud se puede evaluar a partir de conocimientos de historia demográfica. Si se trata de una isla pequeña, alejada de las demás, y sabemos que ha recibido pocos inmigrantes, la fuerza de la deriva puede explicar la aparición de una rama larga. La inmigración también es importante: si es escasa o nula, la deriva es más eficaz (en este caso también se habla de un fuerte aislamiento genético). Si la inmigración es fuerte, la deriva puede desaparecer. Hay muchos ejemplos de ello. La isla de Pascua está muy apartada de las demás islas de Polinesia. Su historia demográfica es conocida, aunque con lagunas y dudas; se sabe que siempre tuvo una población modesta y que en el siglo XVIII hubo un estrangulamiento demográfico. Tiene una rama más larga que todas las demás islas polinesias. Cerdeña es la isla mediterránea más alejada de las costas continentales y ha tenido una historia de largo aislamiento genético. También en ella encontramos los resultados de una fuerte deriva genética. Lo mismo se puede decir de Islandia, aunque genéticamente no está tan aislada del resto de Europa como Cerdeña, porque fue poblada en una época mucho más reciente (en el siglo IX de nuestra era) a partir de un número considerable de colonizadores (quizá 20 000). Todas estas islas tienen ramas de una longitud razonablemente justificada por su historia.
La insularidad geográfica no es el único factor que determina ramas evolutivas largas. Por razones culturales, ciertas poblaciones (como los vascos, los judíos, los esquimales y otros muchos) tienden a limitar su exogamia, es decir, se casan sobre todo dentro de su grupo. Además, en un matrimonio con una persona de una etnia distinta, es más fácil asistir a la pérdida de la identidad cultural de la pareja, que dejará de formar parte de la población. En estos casos de insularidad que podríamos llamar cultural puede haber ramas largas, sobre todo si el grupo es pequeño. Las dos situaciones —pequeñez de la dimensión geográfica y reducción o falta total de matrimonios con los vecinos— tienen el efecto de alargar las ramas.
Las ramas cortas, naturalmente, tienen el origen contrario (una dimensión geográfica grande, si la causa es la deriva), pero puede haber otra causa, lo contrario del aislamiento genético, es decir, una situación de mezcla genética superior a lo normal, que puede acortar una rama. Estas mezclas pueden ser frecuentes cuando alguna migración importante acerca grupos que han estado separados mucho tiempo. La migración (forzosa) de los africanos, llevados como esclavos a América, produjo frecuentes mezclas entre negros y blancos, y entre negros e indígenas americanos. Las mezclas de negros y blancos se reconocen con facilidad, y en Norteamérica los descendientes de uniones mixtas se han clasificado socialmente como negros. Por eso encontramos que los negros norteamericanos han recibido un importante flujo genético de blancos norteamericanos (valorado en un 30 por 100 por término medio, mediante estudios con marcadores genéticos), que es mayor en el norte de Estados Unidos (50 por 100), mientras que en el sur baja hasta el 10 por 100. Se ha calculado que el flujo se debe a una introducción del 5 por 100 de genes de blancos por generación, suponiendo que haya sido constante a lo largo del tiempo, es decir, en los tres últimos siglos, después de la llegada de los esclavos africanos. He aquí un ejemplo (de los muchos que se conocen) de flujo genético.
El árbol de la figura 2 obtenido con los RFLP muestra una rama muy corta un poco inesperada: la de los europeos. Parte casi del mismo centro, y termina cerca de su origen. En este caso no es posible que la población haya sido tan numerosa. La interpretación más sencilla es que los europeos también sean una mezcla genética. ¿Cómo es posible? Si Gobineau —el teórico del racismo del siglo pasado— levantara la cabeza, se moriría de rabia y de vergüenza. ¡El que creía que los europeos, sobre todo los de Europa central, situados en el centro genético de los demás europeos, son la raza más pura genéticamente y mejor dotada psicológicamente y en todos los aspectos, y que pensaba que la mezcla debilitaría y corrompería inevitablemente la raza!
Hoy disponemos de un método para demostrar de un modo riguroso lo que acabo de decir, pero sólo podremos usarlo más adelante, cuando hayamos acumulado datos de otra naturaleza que serían difíciles de confirmar aquí. En todo caso, no cabe la menor duda de que los europeos son casi exactamente intermedios entre los africanos y los orientales. Si buscamos porcentajes más exactos, hallamos que la mezcla parece formada por dos terceras partes de poblaciones de Extremo Oriente y una tercera parte de poblaciones de origen africano. ¿Cuándo se produjo esa mezcla? Los datos señalan una fecha bastante antigua, del orden de 30 000 años atrás. ¿Cómo podemos estar seguros de ello? En los análisis históricos nunca hay nada seguro; nos gustaría apoyamos en datos arqueológicos, pero por desgracia no existen. De todos modos, esto no se puede considerar una prueba negativa, porque sabemos muy poco, por no decir casi nada, de los movimientos de las poblaciones humanas en los últimos 100 000 años.
Pero tenemos que añadir que una hipótesis totalmente contraria también podría explicar la misma observación. Si no hubiera habido ninguna evolución de los europeos —si hubieran permanecido idénticos a los seres humanos modernos de hace 100 000 años—, tendríamos una situación semejante. ¿Sería una suerte o una desgracia, para los europeos, no haber evolucionado nada en los últimos 100 000 años? Para Gobineau sería una gran suerte, porque pensaría que las otras razas se han echado a perder al diferenciarse de los europeos, mientras que éstos todavía son perfectos.
Lo sentimos por Gobineau y los que todavía hoy piensan como él, ya que los datos no nos llevan en esa dirección. Ante todo, existe otro detalle que aún debemos examinar, para excluir que la causa de este fenómeno sea un artificio. Casi todos los polimorfismos clásicos, y todos los del ADN que hemos examinado hasta ahora, siempre se obtuvieron mediante el examen de muestras de genes cuyos polimorfismos se descubrieron en el laboratorio, en Europa o Norteamérica, con muestras de sangre procedentes de europeos o individuos de origen europeo. ¿Pudo generar este eurocentrismo, debido a circunstancias involuntarias, el artificio de colocar las poblaciones europeas en el centro del mundo? La respuesta es sí, pero un examen más profundo demuestra que esto no explica todo el problema, y tal vez ni siquiera la mayor parte.
Es verdad que los análisis de las migraciones de las que hablaremos en el capítulo siguiente han revelado que una parte importante de nuestros genes procede de Oriente Próximo, pero genéticamente esta región del mundo no es muy distinta de Europa. Es verdad que los hunos (una tribu de mongoles) llegaron a Francia e Italia hacia el 450 de nuestra era. Es verdad que los turcos llegaron hasta la frontera de Austria hace tres siglos. Efectivamente, la distribución geográfica de los genes en Eurasia nos muestra una gradación de las frecuencias génicas muy regular de este a oeste. Es difícil decir cuándo tuvieron lugar esas migraciones, pero la gradualidad genética desde el oeste de Europa hasta el este de Asia debe ser el resultado de migraciones numerosas, en ambos sentidos y en épocas distintas.
No nos entusiasman los métodos de máxima parsimonia, que a mi entender han gozado de un favor totalmente injustificado entre los cladistas, pero el análisis cladístico es importante cuando nos permite utilizar razonamientos basados en la identidad de nuestros antepasados, en el paso de genes de ellos a nosotros, y en las consideraciones que permiten a propósito de la elección de los caracteres más útiles para la reconstrucción de la historia genética. Eso es lo que se ha propuesto la cladística moderna, y vale la pena poner un ejemplo.
La mayoría de los RFLP utilizados por nosotros sólo mostraban dos formas del gen (dos alelos) en nuestras poblaciones. El número de las formas de un gen que hay en una población depende directamente de la frecuencia de mutación de dicho gen, que en nuestro caso debe ser pequeña. Esto nos ayudó en nuestro trabajo a identificar, en los monos más cercanos a nosotros (los primates), la existencia de las mismas formas de un gen presente en el hombre. Una frecuencia de mutación alta, sin duda, lo habría impedido.
El resultado fue muy claro. En nuestros primos casi nunca se encontraba el mismo polimorfismo (sólo en uno de 78 genes en el chimpancé). Pero en el chimpancé, el primate más cercano a nosotros, se encontraba con frecuencia una de las dos formas (para el 80 por 100 de los genes estudiados). Por lo tanto, los polimorfismos humanos estudiados por nosotros aparecieron probablemente después de la separación entre el hombre y el chimpancé, es decir, en los últimos cinco millones de años, y sólo en la especie humana. Se puede objetar que el polimorfismo ya podía estar presente en el antepasado común de ambos, pero se perdió en la línea evolutiva que ha llevado hasta el chimpancé en los cinco millones de años transcurridos hasta hoy. La respuesta es negativa, de acuerdo con lo que hemos visto en los primos más lejanos. En efecto, en el gorila y el orangután, que se separaron antes del antepasado común del hombre y el chimpancé, se ha encontrado un porcentaje del 40 por 100 de genes con el tipo humano ancestral (el mismo que se encuentra en el chimpancé), pero nunca los tipos presentes en el hombre y ausentes en el gorila. La presencia del tipo humano ancestral en un porcentaje inferior en el gorila y el orangután era de esperar, dado que se separaron de la línea que lleva al hombre en una época anterior.
Gracias al conocimiento de las formas ancestrales de los genes estudiados hemos podido hacer otras indagaciones. Por ejemplo, hemos podido averiguar cuál era la frecuencia de los genes ancestrales, y su probable edad, en las distintas poblaciones humanas estudiadas. Según una teoría matemática que permite prever el número de las formas mutantes de una edad determinada, cuanto más reciente es la mutación que las ha producido, más pequeña es su frecuencia en la población; las mutaciones más antiguas suelen ser raras.
Este trabajo puso de manifiesto que la población europea tiene una distribución de la edad de las mutaciones completamente distinta de la de las otras ocho poblaciones examinadas. La anomalía es que las formas más comunes de los genes mutantes (no ancestrales) tienen una frecuencia cercana al 50 por 100 en la población europea, mientras que las otras poblaciones siguen la regla que establece la teoría de la edad de mutación: las más comunes son las de menor frecuencia, y más recientes. Esto no tiene nada de extraño, dado el eurocentrismo de la investigación de este polimorfismo. Nosotros mismos, en investigaciones pasadas, buscamos polimorfismos de RFLP y seleccionamos constantemente los que tenían una frecuencia parecida de las distintas formas de un gen, más cercana por tanto al 50 por 100.
El estudio del ADN mitocondrial ha despertado mucho entusiasmo, y en los últimos años su evolución se pudo estudiar mejor que la de los genes cromosómicos. Las mitocondrias son orgánulos presentes en todas las células de los organismos superiores, a veces en decenas de miles, que se ocupan de la producción energética mediante los procedimientos más eficaces. Se cree que al principio eran bacterias que se unieron en simbiosis indisoluble a las células, y se reproducen independientemente del núcleo celular, aunque bajo su control. En el momento de la fecundación parece que sólo la madre transmite sus mitocondrias al hijo, de modo que la transmisión es puramente matrilineal. Una mitocondria posee numerosas copias de un solo cromosoma, de forma circular como los de las bacterias. Este hecho confirma que, en efecto, las mitocondrias son bacterias que empezaron a vivir en simbiosis con células de organismos superiores hace 1000 millones de años o más. Ahora la simbiosis es imprescindible para los dos: el patrón (la célula animal) y el huésped (la mitocondria). El cromosoma mitocondrial es muy corto. También está formado por ADN, con 16 000 nucleótidos, es decir, más o menos la ducentomilésima parte de los que hay en el conjunto de los cromosomas nucleares. En el ADN mitocondrial las mutaciones, por término medio, son diez veces más frecuentes que en los cromosomas nucleares, sobre todo en un segmento especialmente variable.
La transmisión a través de un solo progenitor evita una complicación que entorpece el estudio de los cromosomas del núcleo celular, a saber, que cada individuo recibe una dotación de los dos progenitores (salvo en el caso del cromosoma Y, que sólo es transmitido por los machos y sólo se encuentra en los machos). En el caso del núcleo, las dotaciones del padre y de la madre se mezclan, y se produce el fenómeno conocido como recombinación, un intercambio de elementos entre ambas dotaciones, cuando éstos pasan a las generaciones sucesivas. En el caso de las mitocondrias no sucede lo mismo. El cromosoma mitocondrial se comporta como un segmento genéticamente rígido, y todos los genes que lo forman son transmitidos como un bloque único a las generaciones sucesivas. Esto permite hacer un árbol evolutivo de todas las mutaciones que observamos al comparar distintos individuos; en la práctica se hace un árbol genealógico de los individuos. Hasta hace poco, con los genes cromosómicos, no se podía hacer esto (hoy se ha logrado). Naturalmente, también se pueden construir árboles de poblaciones, como hemos hecho hasta ahora con los genes cromosómicos.
Hay cierta diferencia entre los dos procedimientos de análisis. Lo veremos con una anécdota. Hace unos años me llevé la sorpresa de ser entrevistado por la revista femenina Vogue, porque los científicos hablaban de la fecha de nacimiento de Eva, fijada por Allan Wilson en 190 000 años atrás, con un margen de error comprendido entre los límites de 150 000 y 300 000 años. Era natural que Vogue se interesara por semejante noticia, pero resultaba extraño que los científicos recurrieran a Eva. ¿Por qué hablar de esta mítica figura? Hay un motivo: al retroceder en el tiempo, se puede llegar a un solo antepasado del que descienden todos los tipos de ADN mitocondrial que existen hoy en el mundo. Es el antepasado común más reciente de todos los seres vivos; pero como las mitocondrias son transmitidas, si se puede decir así, sólo por las mujeres, tenía que ser una de ellas, y era bastante natural llamarla Eva en tanto que primera mujer. Pero tenemos que puntualizar que sólo es antepasada en lo que a las mitocondrias se refiere, por lo que para evitar confusiones también ha recibido el nombre de Eva mitocondrial.
La fecha en la que vivió esta Eva se ha calculado con arreglo al número de mutaciones que separan a los hombres de los chimpancés, por un lado, y por otro al que separa por término medio a los africanos de los no africanos. La proporción entre ambos es aproximadamente de 26. Conociendo la fecha en que los hombres se separaron de los chimpancés, hace unos 5 millones de años, se obtuvo que la de la separación entre africanos y no africanos es 26 veces más pequeña, o sea, 190 000 años. (Confieso que he modificado la verdadera relación numérica para no tener que dar demasiadas explicaciones bastante complicadas).
Se suele cometer un error en la interpretación de este análisis. Varios paleoantropólogos creyeron, y puede que alguno aún lo siga creyendo, que los datos genéticos indican que realmente hubo, en un momento dado, una sola mujer, como sugiere el nombre de Eva. Dado que ya hace bastantes años que se habla del origen africano del hombre moderno, les pareció natural poner las dos cosas juntas y hablar de la Eva africana. La expresión tuvo una gran fortuna periodística. La verdad es que cuando la revista Vogue me llamó por teléfono para entender la historia de la Eva africana, yo no tenía la menor idea del asunto, porque aún no conocía el trabajo de Wilson y sus colaboradores. Pero intuí el carácter de la observación y del error, y Vogue (edición norteamericana) publicó mi explicación, que resultó ser correcta. He publicado mis artículos en muchas revistas científicas, y artículos de divulgación científica en varios periódicos y revistas, pero estoy orgulloso de mi contribución a la revista de moda más famosa.
Es importante subrayar que a partir de estos datos no se puede llegar a la conclusión de que hubo un tiempo en que la población humana quedó reducida a una sola mujer, o que hubo un estrangulamiento demográfico de la población humana en la época de la llamada Eva. Entre otras cosas, la fecha de nacimiento de la mujer en la que se produjo la mutación que nos permite fechar el último antepasado (antepasada) común de los africanos y sus descendientes que poblaron el resto del mundo, no coincide con la de la separación entre las poblaciones africanas y no africanas. El nacimiento de la pseudo Eva tiene que ser anterior, un número de años desconocido, al de la separación de las poblaciones. Efectivamente, la fecha de 190 000 años para la pseudo Eva es más antigua que la de 100 000 años de la separación entre africanos y no africanos, según los datos arqueológicos disponibles.
Se ha hablado mucho acerca de la Eva africana. Muchos investigadores han arremetido contra la fecha y la interpretación. Eva africana es el producto de una importante serie de estudios del laboratorio de Allan Wilson, de Berkeley. Por desgracia, a Allan nos lo arrebató una leucemia aguda en 1991, y la polémica se avivó después de su muerte.
Pero no vale la pena que nos detengamos en las críticas al trabajo y las conclusiones de Allan Wilson, porque una investigación japonesa muy reciente confirma las conclusiones anteriores y proporciona una estimación más sólida de la fecha del nacimiento de Eva.
Horai et al. analizaron la secuencia completa en el cromosoma mitocondrial de tres hombres (un africano, un europeo y un japonés) y la compararon con las secuencias de cuatro primates: dos especies de chimpancé, el gorila y el orangután. Los resultados se ilustran en la figura 3. La fecha de esta Eva es de 143 000 años, con un margen de error bastante reducido. La separación entre el hombre japonés y el europeo, naturalmente, es más tardía. Las fechas de separación de los otros primates corresponden a los resultados anteriores. Esta investigación es la más segura y completa que se ha realizado hasta hoy sobre el ADN mitocondrial. Naturalmente, nos gustaría tener datos sobre los cromosomas del núcleo celular, que poseen una cantidad mayor de información, una frecuencia de mutación distinta y otras particularidades cuya importancia en relación con nuestro problema no somos capaces de apreciar.
Puede que la espera para conocer esta respuesta haya sido larga, pero hemos tenido suerte.
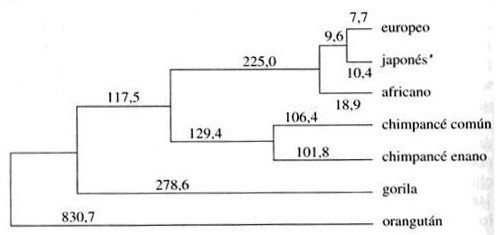
3. Análisis del ADN mitocondrial en tres seres humanos y en otros cuatro primates (según Horai et al., 1995).
La genética molecular moderna, y en particular el análisis del genoma humano, han dado lugar a muchas novedades. Una bastante sorprendente es que los cromosomas (y no sólo los humanos) contienen gran número de repeticiones en tándem de algunas secuencias. Nos ocuparemos de las que resultan más útiles para nuestros estudios, las de secuencias muy cortas, de dos a cinco nucleótidos. Las más corrientes están formadas por los nucleótidos citosina y adenina, CA. En segmentos de ADN de más de diez o quince repeticiones CACACACA… hay muchas posibilidades de que el proceso de duplicación del ADN tenga un ataque de tartamudeo y la secuencia se alargue, o se acorte. El cambio más corriente es la suma o la sustracción de un grupo CA.
Cuando la frecuencia de mutación es alta, se encuentran muchas formas de uno de estos segmentos de ADN en distintos cromosomas, que se diferencian en el número de repeticiones: por ejemplo, 11, 12, 13, 14 y 15. Un individuo heterocigoto tiene dos formas, por ejemplo 12 y 15. El análisis en el laboratorio de esta variación es bastante rápido. La gran ventaja es que en el genoma hay muchos tartamudeos de este tipo, y cada uno genera una variación entre individuos, que puede servir de marcador genético. Dada la necesidad de marcadores genéticos, numerosos laboratorios han identificado los lugares de los cromosomas donde se encuentran los tartamudeos. La Genethon ha sido especialmente activa y ha aislado más de 6000, quizá el 10 por 100 de todos los que existen en el núcleo humano. Es muy fácil trabajar con estos marcadores, llamados microsatélites. Están repartidos casi al azar por todo el genoma. Se cree que por término medio hay uno cada 50 000 nucleótidos.
La aplicación evolutiva más interesante de los microsatélites es un método de datación genética absoluta. Podemos comparar fácilmente la diferencia media entre el número de repeticiones de dos poblaciones. Según una sencilla teoría matemática, esta diferencia tiende a crecer con el aumento del tiempo de separación entre dos poblaciones. Exactamente, la diferencia (elevada al cuadrado) es igual a 2 multiplicado por el tiempo medio de separación entre las dos poblaciones (calculado en número de generaciones), multiplicado por la frecuencia de mutación por generación. Casi todos los microsatélites que hemos estudiado son del tipo CA, y su frecuencia media de mutación es conocida. Resulta sencillo, pues, calcular la fecha de la separación entre africanos y no africanos, o sea, la fecha en que la primera población africana salió de África para asentarse en Asia. El resultado es unos 80 000 años, de acuerdo con los resultados más recientes calculados con casi 100 microsatélites. Como era de esperar, es inferior al que se ha calculado muy recientemente para la Eva africana. Esta estimación todavía tiene un elevado error estadístico, que se podrá reducir estudiando muchos otros microsatélites. Es sólo cuestión de trabajo.
Lo bueno de este método de datación es que no recurre a un tiempo exterior de referencia, como en los otros métodos mencionados hasta ahora (por ejemplo, en el caso del ADN mitocondrial, la base de referencia era el tiempo de separación entre el chimpancé y los hombres). Es un método independiente, que no está sujeto a error si la fecha de referencia está equivocada. Se puede decir, por lo tanto, que se trata de un método de datación absoluta, porque sólo utiliza la información genética y no necesita la paleontológica. El reloj utilizado es la frecuencia de mutación. Es la que marca el tiempo; y el que ha transcurrido desde la separación de las poblaciones se mide por el número de mutaciones que las diferencian.
Una datación absoluta no quiere decir que el reloj sea necesariamente perfecto. El radiocarbono permite una datación absoluta. Su radiactividad disminuye con una velocidad conocida: cada 4800 años se reduce a la mitad, y en él se basa la estimación de la cantidad relativa del carbono radiactivo con respecto al carbono no radiactivo presente, que permanece en una muestra de materia vegetal y nos dice lo antigua que es. Pero el método se basa en una hipótesis fundamental: que la cantidad de carbono radiactivo presente en la atmósfera ha permanecido constante desde hace mucho tiempo. Los estudios realizados determinando la edad, ya conocida, de maderas muy antiguas, por medio de la dendrocronología (recuento de los anillos del tronco) y la cantidad de carbono radiactivo presente, han demostrado que la hipótesis no siempre se cumple, y han permitido aportar una corrección. Ahora que hemos introducido este nuevo método de datación genética, nosotros también podremos controlar los posibles errores introducidos, por ejemplo, si las frecuencias de mutación no son constantes de unas poblaciones a otras. Hoy por hoy, en todo caso, resulta muy alentador que este método dé prácticamente los mismos resultados que se obtienen con el análisis del ADN mitocondrial, aunque tenga una base teórica muy distinta.