Un paseo por la selva
Hace muchos años me planteé el problema de si sería posible reconstruir la historia de la evolución humana a partir de los conocimientos genéticos sobre las poblaciones vivientes. Hasta entonces el estudio de la evolución había sido sobre todo una historia de cráneos fósiles, y no había llevado muy lejos. El material paleoantropológico no es muy rico, e incluso hoy debemos contentarnos con un número bastante reducido de muestras fósiles. Por eso, el número de piezas es tan reducido que la reconstrucción total del rompecabezas puede parecer una meta inalcanzable. No es de extrañar, pues, que el hallazgo de un nuevo fósil o la revisión de una fecha puedan tener consecuencias sorprendentes, que ponen patas arriba las conclusiones aceptadas hasta entonces. Esto explica, en parte, la atención prestada por la prensa a las novedades sobre el pasado humano. Los conocimientos que tenemos son tan limitados que el descubrimiento de media mandíbula con un millón de años de antigüedad ocupa páginas enteras en los periódicos. Y la avidez por nuevos restos que hablen de nuestro pasado es grande.
Si dejamos a un lado cráneos y esqueletos, encontramos en la genética un material que proporciona muchos más datos, aunque hasta ahora limitado casi exclusivamente a las poblaciones vivientes. Otra ventaja frente al material paleoantropológico es que los datos genéticos (ya sean individuales o de poblaciones) varían en el tiempo con arreglo a unas leyes conocidas y precisas. La evolución de los huesos también depende de la del material genético, pero de un modo más complicado y menos conocido. Además, en ella intervienen directamente otras condiciones que no tienen nada que ver con la herencia genética.
Hasta hace pocos años no podíamos fiarnos de los análisis de caracteres genéticos realizados con muertos, por lo que no había esperanzas de obtener informaciones del mismo tipo sobre los hombres más antiguos, para tener también datos genéticos fósiles que se puedan comparar con los modernos. Pero en fechas muy recientes se ha comprobado que a veces el ADN[8] también puede conservarse de un modo sorprendente. El ADN es el material genético por excelencia, por lo que es ideal para los estudios sobre la evolución. Los mejores resultados sobre el ADN antiguo se han obtenido con insectos y hojas incluidos en el ámbar, con una antigüedad de decenas de millones de años. El ámbar es resina solidificada, y la conservación en ámbar es excelente, pero nunca encontraremos dentro un elefante o un hombre. El novelista Michael Crichton ha dado gran popularidad, en Parque jurásico, a la idea de devolver la vida a los dinosaurios utilizando el ADN contenido en los cadáveres de moscas y mosquitos conservados en ámbar, que los habían picado y habían chupado su sangre. Hay que decir que, aunque la genética molecular está muy desarrollada, cuesta imaginar que algún día se pueda llegar a estos resultados. Una vez me acusaron de haber recreado un hombre de Neandertal con el mismo método, pero creo que semejante hazaña, que desde luego no recomiendo por razones éticas, sólo es posible en la imaginación de los escritores.
Recientemente se encontró un hombre de la Edad del Bronce sepultado en los hielos de los Alpes orientales. Le llamaron Oetzli, por el nombre del lugar donde se había encontrado. Oeztli ha proporcionado informaciones muy valiosas sobre los vestidos y los utensilios que llevaba, pero las obtenidas a partir del estudio de su ADN han sido menos emocionantes; se diría que la conservación en hielo no es tan buena como en ámbar. Ya tendremos ocasión de hablar del ADN mitocondrial, mejor conocido que el ADN nuclear por dos razones: es 200 000 veces más corto, y la secuencia de sus nucleótidos[9] se conoce por completo. La segunda razón es que la mayoría de las células sólo tienen dos copias de ADN nuclear, pero decenas de miles de copias de ADN mitocondrial, por lo que es más fácil encontrar alguna intacta en los restos muy antiguos, si están bien conservados. Se ha podido reconstruir la secuencia de un centenar de nucleótidos en una región del ADN mitocondrial de Oetzli, llamada D-loop, que es muy variable de unos individuos a otros. El hombre tirolés de la Edad del Bronce ha resultado ser similar a los hombres modernos de la región, algo que está de acuerdo con el hecho de que no ha habido inmigraciones importantes de lugares lejanos desde la Edad del Bronce hasta hoy.
Así pues, el análisis del ADN de los hombres antiguos aún tiene muchas limitaciones. Está sujeto a las mismas probabilidades que condicionan todos los hallazgos fósiles: son pocos, dependen del azar, y la edad de los restos a veces es difícil de establecer, sobre todo si supera los 40 000 años. En efecto, con el mejor método de datación que tenemos, el del radiocarbono, difícilmente se pueden datar objetos más antiguos. Pero esta clase de investigaciones, evidentemente, tiene un gran futuro, pues el ADN puede proporcionar informaciones mucho más seguras que las obtenidas examinando los huesos. Por desgracia, el estado de conservación suele ser bastante malo, a causa de la fragmentación sufrida por el ADN antiguo, lo que hace muy laboriosa y difícil la reconstrucción de la secuencia.
En cambio, tenemos muchos conocimientos sobre las poblaciones modernas, y el desarrollo reciente de la biología molecular nos brinda unas posibilidades casi ilimitadas de seguir ampliándolos. ¿Se pueden utilizar estos conocimientos para reconstruir la evolución humana, y cuáles son los problemas que esperamos resolver? Cuando empecé a pensar en ello trabajaba en la Universidad de Cambridge, en Gran Bretaña, en el departamento de genética dirigido por el profesor R. A. Fisher. Me ocupaba de la genética de las bacterias, y sólo a partir de 1951, cuando empecé a dar clases a tiempo parcial en la Universidad de Parma, tuve ocasión de ocuparme de la genética de las poblaciones humanas. Mi metamorfosis en genetista humano fue bastante lenta, y no fue hasta 1961 cuando decidí que había llegado el momento de tratar de resolver el problema específico de reconstruir la evolución humana a partir de los datos genéticos modernos. Mientras tanto había tenido la posibilidad de desarrollar alguno de los métodos que se necesitan para este fin.
Después de Darwin, la evolución se solía representar por medio de árboles, para indicar el modo en que se formaron las especies, unas a partir de otras (definición de especie: véase cap. 1, nota 7, p. 209). Entre todas las poblaciones humanas la fecundidad es completa y sin limitaciones, lo que nos permite concluir no sólo que todos pertenecemos a la misma especie, sino también que la separación entre grupos humanos debe ser muy reciente, pues de lo contrario notaríamos una disminución de la fecundidad entre individuos de grupos distintos, es decir, el principio de la formación de especies distintas.
Si en el transcurso de la evolución humana ha habido separaciones bastante claras entre dos poblaciones, como debió suceder por ejemplo cuando de una población madre se desgajó una colonia y los intercambios entre población madre e hija escasearon o se interrumpieron, el modelo de las colonizaciones sucesivas se puede comparar con el de las ramificaciones de un árbol.
Cada ocupación de un nuevo continente debió de ser consecuencia de, por lo menos, un episodio de colonización de este tipo. El paso de un continente a otro tiene casi necesariamente el carácter de un fenómeno discontinuo. Aunque el paso requiere períodos largos, y se ve favorecido por una continuidad geográfica temporal, es muy probable que haya habido discontinuidades del fenómeno en el tiempo. Veamos el ejemplo de América: hace 25 000 años o más se formó un puente entre Siberia y América del Norte, ya que el nivel de los mares había descendido a causa de la congelación de una parte del agua marina, transformada en hielo como consecuencia del enfriamiento de la última era glacial. La tierra intercontinental que entonces unía Asia con América del Norte se ha llamado Beringia. Cuando el clima volvió a ser cálido, hace 10 000 años, parte del hielo polar se fundió, y las aguas volvieron a cubrir Beringia, formando el actual estrecho de Bering. En el período de tiempo comprendido entre 25 000 (o quizá más) y 10 000 años atrás, debió de ser bastante fácil pasar de Asia a América, pero con la baja densidad de población y las difíciles condiciones climáticas de la época las migraciones serían de grupos reducidos. Las que se produjeron después de la desaparición de Beringia se hicieron en barco, y fueron aún menos numerosas. La última fue la de los esquimales, que procedían del norte de Asia y ocuparon el extremo norte de América. Probablemente hubo pocos intercambios entre los esquimales y los inmigrantes que les habían precedido. La situación lingüística entre los indígenas americanos indica que hubo pocas migraciones desde Asia, quizá sólo tres[10].
Si queremos comparar las poblaciones indígenas de los cinco continentes desde un punto de vista genético, lo más sencillo es calcular las distancias genéticas entre continentes, examinándolos de dos en dos y hallando las medias de las distancias observadas para muchos genes (en este caso disponemos de un centenar). Los resultados de los cálculos se resumen en el siguiente cuadro.
Genéticas entre continentes
| África | Oceanía | América | Europa | |
| Oceanía | 24,7 | |||
| América | 22,6 | 14,6 | ||
| Europa | 16,6 | 13,5 | 9,5 | |
| Asia | 20,6 | 10,0 | 8,9 | 9,7 |
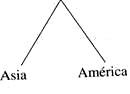
A partir de estas distancias, ¿cómo podemos construir un árbol que nos ilustre la historia de las separaciones sucesivas que han dado lugar a estas diferencias? Actualmente existen varios métodos, por lo general bastante complicados. Voy a explicar uno sencillito, aunque no es el que utilizamos la primera vez. Comparemos los cinco continentes ya colocados, en el cuadro anterior, en un orden determinado (como quedará más adelante). Ante todo se busca la distancia menor, que es la que hay entre americanos y asiáticos. Entonces suponemos que esta separación es la última en orden cronológico. Esto equivale a aplicar el principio de que la distancia genética entre dos poblaciones aumenta al hacerlo el tiempo de separación entre ellas. Este principio es razonable, pero no necesariamente verdadero; de todos modos nos basaremos en esta hipótesis y trataremos de verificarla a continuación con otros datos. Pero hay que destacar que las diferencias entre las distancias que aparecen abajo a la derecha en el cuadro (9,5; 8,9; 9,7) son muy pequeñas. Por otros cálculos sabemos que el error estadístico en este caso es del orden del 20 por 100, de modo que la elección de la distancia menor entre las tres es, por así decirlo, arbitraria. Pero de momento pasaremos por alto este problema y admitiremos que, según los datos del cuadro, la distancia más pequeña (8,9), y por lo tanto la división más reciente, es entre Asia y América. De modo que empezaremos a construir así el árbol:
Ahora podemos reunir los dos continentes, hallando la media de las distancias de cada uno de los demás con Asia y América. Por ejemplo, la media entre Europa y Asia (9,7) y Europa y América (9,5) es 9,6. El cuadro de partida pierde una línea y una columna, y se convierte en:
| África | Oceanía | Asia-América | |
| Oceanía | 24,7 | ||
| Asia-América | 21,6 | 12,3 | |
| Europa | 16,6 | 13,5 | 9,6 |
Busquemos de nuevo la distancia más pequeña: es la que hay entre la media de Asia y América y Europa. Añadimos otra rama al árbol uniendo Asia-América con Europa:
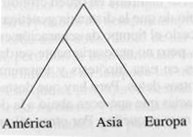
Podemos seguir; el paso siguiente añade Oceanía, y el último África.
El árbol será, pues:
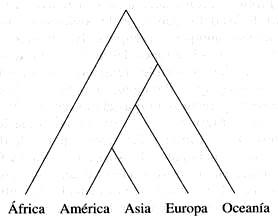
Más adelante veremos que un modelo de grandes migraciones que, partiendo de África, llegaron primero a Australia, luego a Asia, luego a Europa y a América, está de acuerdo con estas distancias, y también está de acuerdo con los datos arqueológicos. Nuestro árbol tiene bastantes posibilidades de representar la evolución del hombre moderno.
Lo que nos interesa no es la clasificación de las razas[11], entidades que escapan a una definición precisa, sino reconstruir el árbol evolutivo o filogenético de las poblaciones humanas. El gran clasificador de las plantas y los animales, Linneo, trabajó antes de que apareciera el concepto de evolución. Como casi todos sus contemporáneos, creía que las especies vivas habían sido creadas tal como son. Se pueden hacer muchas clasificaciones del mismo material, y la introducción del ordenador ha hecho posible la llamada taxonomía numérica, que facilita el trabajo del clasificador. Pero los taxonomistas numéricos que elaboraron métodos de clasificación automática no se plantearon problemas evolutivos: buscaban clasificaciones de valor general, un concepto difícil de definir. Nosotros optamos por desarrollar métodos basados en la teoría de la evolución biológica, y el primero de ellos data de 1963. Se basa en el principio de que en el proceso evolutivo una población inicial a menudo se divide en fracciones que ocupan regiones distintas y evolucionan de forma independiente, diferenciándose entre sí a medida que pasa el tiempo desde la separación. El proceso prosigue, generando el árbol evolutivo.
Nuestra primera reconstrucción filogenética de la evolución humana a partir de los datos genéticos de poblaciones indígenas modernas fue un éxito, aunque no completo. El árbol reconstruido con ordenador reunió las poblaciones del mismo continente en grupos continentales, revelando posibles relaciones recíprocas de origen entre continentes o grupos. Por ejemplo, indica que los amerindios son genéticamente parecidos a los asiáticos del este, y en efecto, de acuerdo con otros criterios y datos antropológicos, se les considera primos lejanos suyos. También descubre el parecido de los asiáticos del este con los habitantes de Australia y Nueva Guinea. Los europeos aparecen en una posición intermedia entre los africanos y el resto del mundo. Esta posición intermedia ha dificultado durante mucho tiempo el reconocimiento de la primera ramificación en el árbol evolutivo humano. Al principio parecía que separaba a los africanos y europeos del resto del mundo, pero con los datos más recientes está claro que los africanos fueron los primeros en separarse de todos los demás, reforzando la hipótesis de que el hombre moderno nació en África y luego se difundió por toda la Tierra. De modo que el éxito de nuestro trabajo no fue completo, pero casi. Había complicaciones difíciles de entender hace treinta años. Con nuestros actuales conocimientos, podemos decir que los genes examinados no eran suficientes, pero hay algunos motivos más.
Varios métodos distintos de construcción de los árboles pueden dar resultados algo distintos con el mismo grupo de frecuencias génicas. Se han elaborado muchos otros métodos, y cada uno da resultados ligeramente distintos de los demás. Vale la pena explicar una de las razones de esto. La principal dificultad es que los árboles por elegir son numerosos. En efecto, su número crece con una velocidad vertiginosa si aumenta el número de poblaciones estudiadas. La situación más sencilla es aquella en la que examinamos tres poblaciones. Si queremos localizar la raíz, debemos elegir entre tres árboles:
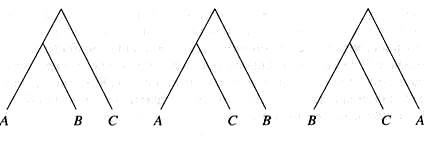
Si tomamos cuatro especies, los árboles posibles son quince. Si nos olvidamos de la raíz, aún quedan tres árboles:
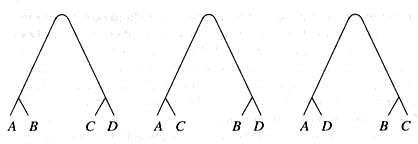
(Dado que no se especifica la raíz, la indicamos redondeada). Con cinco poblaciones los dos tipos de árboles son 105 (con raíz) y 15 (sin raíz); con diez poblaciones, 34 millones y 2 millones. Con veinte poblaciones el número de árboles es más o menos un 8 seguido de 21 ceros, y 37 veces más pequeño si no tenemos en cuenta las raíces. Para estar seguros de encontrar el árbol mejor, de acuerdo con el criterio utilizado por el método de análisis que hemos decidido emplear, en teoría habría que examinar todos los árboles posibles (con la excepción de un solo método de análisis). Si no queremos renunciar a un análisis completo de todos los árboles posibles, nos resultará difícil, incluso con los ordenadores modernos, tomar en consideración más de una docena de poblaciones.
La situación es aún peor si se quiere utilizar el método más satisfactorio de acuerdo con los dictámenes de la estadística moderna, el de la máxima verosimilitud. Tiene un solo defecto, que los cálculos son muy largos, incluso cuando se utilizan potentes ordenadores. Su gran ventaja es que permite abordar de forma muy rigurosa el problema del análisis de los datos observados. Entonces podemos obtener de un modo completamente satisfactorio una medida de la verosimilitud con que la hipótesis elegida representa nuestros datos. Si se quieren verificar numerosas hipótesis, el método también permite elegir la mejor, porque nos da una medida de la credibilidad de cada una de ellas.
En la aplicación más sencilla del método de la máxima verosimilitud planteamos la hipótesis de que la velocidad evolutiva es la misma en todas las ramas del árbol. En su aproximación, el average linkage, emitimos la misma hipótesis. Pero si no es así, ¿qué podemos esperar? La pregunta nos lleva inmediatamente al estudio de los factores evolutivos, de los que depende la velocidad del cambio de los polimorfismos en el tiempo.
Gracias a la genética moderna aplicada a la teoría de la evolución se han podido conocer cuatro factores evolutivos principales: la mutación, que produce los nuevos tipos genéticos, la selección natural, que elige a los que mejor se adaptan al ambiente en el que viven, la deriva genética, que es el efecto del azar debido a las fluctuaciones estadísticas de las frecuencias génicas de una generación a otra, y la migración de individuos de una población a otra o de un lugar a otro. Empecemos por la deriva genética, el factor más duro de roer, porque generalmente en la enseñanza secundaria no aprendemos ni siquiera los principios del cálculo de probabilidades. Es ni más ni menos que el efecto de las fluctuaciones estadísticas, inevitables en el paso de una generación a otra. Entre los indios de América, casi el 100 por 100 de los individuos es del grupo 0, algo bastante frecuente. Una posible explicación es que los primeros inmigrantes fueran muy pocos, y todos del grupo 0. Supongamos que se originaron de una población en la que había el 50% de individuos del grupo 0, algo bastante frecuente. Si los primeros que emigraron de Asia a América, de los que descendieron los indígenas americanos, eran realmente pocos (digamos una decena o menos), puede que casualmente fueran todos del grupo 0. Con diez emigrantes esta probabilidad es más o menos del uno por mil, pequeña, pero no despreciable. Si no hubo nuevas mutaciones, o migraciones de individuos de otro grupo sanguíneo (A, B o AB), posteriores a la primera colonización, si todos los primeros colonizadores hubieran sido del grupo 0, lo serían también todos sus descendientes. Es un efecto estadístico normal de las dimensiones demográficas de una población. Cuanto más pequeña es la población, mayores son las posibles fluctuaciones aleatorias de las frecuencias relativas de los genes contenidos en los espermatozoides y en los óvulos que formarán la generación sucesiva. Y aunque entre los primeros colonizadores hubiera algunos individuos A y B, su eliminación aleatoria pudo tener lugar en una generación posterior a la primera[12]. Más adelante, cuando el número de individuos que forman la población es grande, todavía se manifiestan las fluctuaciones aleatorias, es decir, la deriva genética, pero son menos importantes.
El nombre de deriva genética dado a las fluctuaciones estadísticas de las frecuencias de los genes puede confundir las ideas, porque parece indicar una tendencia en una dirección determinada, pero la deriva no genera propensión al aumento de uno u otro tipo genético. La única tendencia es hacia la homogeneización de la población, en el sentido de que, si la deriva genética puede actuar libremente sin que las nuevas mutaciones o migraciones introduzcan nuevos tipos genéticos, al final la población estará formada por un solo tipo. Pero si observamos dos poblaciones idénticas, en las que al principio hay varios tipos genéticos, al final de un proceso de deriva genética ambas serán homogéneas, pero tal vez completamente distintas, al estar una de ellas formada por un solo tipo, y la otra por el otro tipo, de los dos que había al principio. A diferencia de la selección natural, la deriva genética es ciega en relación con el resultado evolutivo final, que siempre es la homogeneización completa. Pero hasta entonces la frecuencia de los tipos genéticos de una población aumenta o disminuye, totalmente al azar, en cada generación.
¿Se debe a la deriva la separación de los grupos A y B en América? No se sabe con seguridad, pero también debemos considerar otra hipótesis: es posible que la responsable haya sido la selección natural. A menudo los individuos de grupos distintos tienen una resistencia distinta a ciertas enfermedades. Con el sistema ABO esto se ha comprobado en el caso de numerosas enfermedades infecciosas. Pues bien, parece que la sífilis se propagó por Europa tras el regreso de Cristóbal Colón de América, y se ha sugerido que era frecuente en América y fueron los marineros españoles quienes la llevaron a Europa. En efecto, parece que después de propagarse por España, pasó a Francia y a Nápoles durante las guerras entre Francia y España y la conquista española del sur de Italia, y luego al resto de Europa (por eso también se llamó mal francés y mal napolitano). Hay datos que hacen pensar que también hoy los individuos del grupo sanguíneo o son más resistentes que los demás a esta enfermedad. Además, unas investigaciones realizadas con las momias precolombinas (que, sin embargo, no se han repetido o confirmado con métodos más modernos) hacen pensar que los grupos sanguíneos A y B existían en América hace varios miles de años. Si se confirman estos datos, habrá que optar por una hipótesis de selección natural, según la cual la sífilis fue la causante de la desaparición de los genes A y B en América.
La elección entre las dos interpretaciones posibles, una de ellas basada en la deriva genética y la otra en la selección natural, suele ser difícil, pero siempre es una ayuda saber que la deriva genética influye en todos los genes, aunque en direcciones e intensidades distintas según las leyes del azar, mientras que la selección natural actúa ante todo sobre determinados genes, y en una dirección precisa. Hay situaciones demográficas especiales: pequeño tamaño de la población, o aislamiento prolongado (con escasa o nula inmigración), en las que es probable, a priori, que la deriva genética sea muy activa. En el caso de islas, como Cerdeña, o de poblaciones aisladas, como los vascos (de los que volveremos a tener la ocasión de hablar), se observan desviaciones importantes con respecto al resto de Europa para varios genes. Hay bastantes posibilidades de que en ambos casos, en el pasado, haya habido períodos en que la población era escasa y la inmigración pequeña o nula, unas condiciones en que la deriva genética pudo ser especialmente activa. Pero esto no excluye que algunos de sus genes también hayan acusado la influencia de la selección natural. En Cerdeña tenemos algunos ejemplos clarísimos del aumento de ciertas anemias hereditarias a causa de un factor selectivo, el paludismo.
Las mutaciones son cambios fortuitos en un edificio molecular adaptado a una o varias funciones precisas por una larga historia selectiva. Por eso es poco probable que una mutación, que altera este edificio, sea ventajosa para el organismo. Lo más frecuente es que sea perjudicial, causa de enfermedad o muerte. La selección natural elimina, tarde o temprano, todas las mutaciones nocivas. Pero hay muchas que ni benefician ni perjudican al organismo: son selectivamente neutras. Estas mutaciones tienen más posibilidades de propagarse en una población sometida a la deriva. Si la selección natural es fuerte, el proceso de sustitución de un gen aventajado selectivamente puede durar sólo unos miles de años, como ha sucedido en Europa y parte de África con la capacidad de los adultos para aprovechar el azúcar de la leche (la lactosa). Los niños aprovechan bien la lactosa de la leche, pero pierden esta capacidad a los tres o cuatro años, cuando han dejado de alimentarse con la leche materna. En las poblaciones que aprendieron a criar ovejas, cabras, vacas y otros animales, como los camellos, cuyos adultos siguen alimentándose de leche fresca, ha habido una selección favorable al tipo genético que permite al adulto conservar la capacidad de aprovechar la lactosa. Puede que este proceso selectivo no requiriera más de 5000 o 10 000 años, porque antes no había animales domésticos. Las poblaciones en las que esta costumbre tiene especial importancia son las del norte de Europa y algunas de pastores africanos: en ellas, casi el 100 por 100 de los individuos poseen el nuevo tipo genético que permite a los adultos utilizar la lactosa, un tipo genético que no existe o es escaso en otras poblaciones. Pero por lo general la ventaja de una mutación debida a la selección natural es más modesta, y el proceso selectivo más lento.
En la mayoría de los casos, al final de un proceso selectivo la frecuencia del tipo genético favorito alcanza el 100 por 100, aun partiendo de una frecuencia inicial muy pequeña, quizá de un solo individuo portador de la primera mutación en toda la población. Pero en otros casos el proceso selectivo puede detenerse antes de alcanzar el 100 por 100, y la frecuencia génica puede estabilizarse en un nivel intermedio entre el o y el 100 por 100. La mutación responsable de una enfermedad, un tipo de fibrosis quística más frecuente en el norte de Europa, se pudo producir una sola vez, para luego aumentar en la población, pero el aumento se ha detenido en una frecuencia baja. Si esta enfermedad hubiera podido propagarse a toda la población, estaríamos todos gravemente enfermos, y la población dejaría de existir. Evidentemente esto no ocurre, porque la selección natural ejerce un control automático que mantiene en niveles bajos la frecuencia de la enfermedad.
La selección natural se debe al hecho de que hay diferencias de mortalidad y fecundidad entre los distintos tipos genéticos. Por ejemplo, los individuos afectados de una enfermedad genética (es decir, estrictamente hereditaria) que les mata antes de que puedan reproducirse, no pueden contribuir a la generación sucesiva. Esa enfermedad genética desaparece por completo de la población si no es introducida constantemente por nuevas mutaciones. Si la probabilidad de que estos individuos mueran antes de reproducirse es mayor del 0 por 100, la enfermedad puede alcanzar niveles más altos en la población, que se pueden predecir con sencillas fórmulas matemáticas. La mortalidad no es el único factor, también cuenta la fecundidad. Si se conocen la fecundidad y la mortalidad de los tipos genéticos producidos por un gen, es posible prever matemáticamente el efecto de la selección natural[13].
Es razonable decir que un tipo genético con menos mortalidad (y/o más fecundidad) que otro está mejor adaptado a un medio determinado, se sobreentiende que en ese medio (es raro que un tipo sea mejor que otro en todos los medios posibles). Un tipo así se verá favorecido por la selección natural. Esta consideración lleva a un razonamiento que se suele condensar en la expresión: «La selección natural es la supervivencia del más apto», que si nos descuidamos nos hace caer en una tautología, pues a la pregunta de quiénes son «los más aptos» algunos contestan: «Los favorecidos por la selección natural». En realidad no hay tautología, ya que ciertas condiciones demográficas medibles (véase la nota 6, p. 211), y resumidas en la expresión «valor selectivo y reproductivo de un tipo genético», son las que hacen que un tipo genético se vea o no favorecido por la selección natural. El hecho de que lo llamemos más apto es sólo una cuestión de comodidad.
En el siglo pasado, sobre todo, se daba mucha importancia a la «pureza de la raza». Esto sucede aún con los animales domésticos, ya que al criador le gusta que sus animales sean homogéneos. Los concursos de perros y gatos establecen un ideal de perfección estética (por lo general bastante arbitrario), al que el animal se tiene que acercar todo lo posible. Es un juego biológicamente arriesgado, pues todos los criadores saben muy bien que cuando se persigue la pureza genética de un linaje con cruces repetidos entre parientes consanguíneos, como padre-hija o hermano-hermana, la resistencia a las enfermedades y la fecundidad, junto con otros caracteres deseables, pueden situarse en un nivel peligrosamente bajo.
En general, convendría buscar lo contrario: los animales de cualquier especie, incluido el hombre, tienen más posibilidades de poseer niveles elevados de caracteres importantes, como la resistencia a las enfermedades, la fecundidad, la inteligencia, etc., si son mezclas genéticas. En efecto, se habla de vigor de los híbridos. Si nos referimos a un gen en particular, hablamos de ventaja del heterocigoto, como veremos a continuación.
Conocemos la causa de la ventaja del heterocigoto en muchas situaciones. Los heterocigotos por talasemia (también llamada microcitemia o anemia mediterránea, muy frecuente en algunas zonas de Cerdeña y en Ferrara) son prácticamente normales en el aspecto clínico, pero más resistentes al tipo de paludismo más peligroso. Encontramos frecuencias bastante elevadas de estos heterocigotos allí donde ese paludismo estaba muy difundido. El homocigoto con gen talasémico muere a causa de una anemia que hoy día se puede curar, aunque a veces con grandes dificultades (con muchas transfusiones, o mejor con trasplante de médula ósea sana). Donde no hay paludismo, el gen no supone ninguna ventaja, y casi nunca está presente, pero donde el paludismo era grave, hasta hace poco, todavía hoy se encuentra hasta un 20 por 100 de heterocigotos. La anemia falciforme es otra enfermedad hereditaria cuyos heterocigotos son clínicamente sanos, pero están más protegidos contra el paludismo que los homocigotos normales. Está difundida sobre todo en África, Arabia y la India. Las mutaciones talasémica y falciforme no pueden llegar a ser mayoritarias porque la población ya no lograría reproducirse con normalidad. En la práctica no sobrepasan la frecuencia dictada por un equilibrio delicado entre la ventaja selectiva del heterocigoto y los dos homocigotos.
La selección natural puede ocasionar, pues, variaciones rápidas de la frecuencia génica, tanto más rápidas cuanto mayor sea la diferencia de resistencia y de fertilidad entre dos o más tipos genéticos competidores. Por lo general desemboca en la sustitución de un gen: la forma «vieja», antecedente o ancestral, es sustituida por una forma distinta, «nueva», que ha aparecido en la población a causa de una nueva mutación. Pero en presencia de una ventaja del heterocigoto frente a los dos homocigotos respectivos, la selección natural mantendrá el gen en una condición de equilibrio intermedio entre la desaparición de la forma vieja y su sustitución por la nueva.
En el paso de una generación a otra también interviene la acción del azar, la deriva genética, que encontraremos siempre, incluso en las poblaciones muy grandes, pero se nota sobre todo si la población es pequeña o ha sufrido previamente una «estrangulación» demográfica. La deriva también puede ocasionar la desaparición o el éxito total de un tipo genético, naturalmente dentro de los límites permitidos por la selección natural.
Hasta ahora no hemos hablado de la mutación, salvo para decir que proporciona las novedades, el material sobre el que actúan la selección natural y la deriva. La mutación suele ser un fenómeno raro, pero esto no reduce su importancia, y por otra parte añade otro mecanismo aleatorio a la evolución, pues la manifestación de una mutación puede tener una importancia evolutiva muy grande.
Algunas mutaciones señalan sucesos excepcionales, aunque no siempre los determinan. Probablemente fue una sola mutación lo que determinó el paso de la simetría bilateral (la correspondencia especular entre la mitad derecha y la izquierda), que se encuentra en casi todos los organismos, a la simetría de 3, 4 ó 5 ejes, como encontramos en muchas flores y también en los animales (por ejemplo, los equinodermos). Sabemos que con una sola mutación la mosca clásica de los estudios de genética, la Drosophila, puede pasar de dos a cuatro alas, una modificación que en los insectos separa a los dípteros de los himenópteros. Muy pocas mutaciones tienen efectos tan notables. Pero la suma de muchas mutaciones puede marcar la diferencia entre la salud y una enfermedad determinada.
La aparición de estas mutaciones se puede considerar un fenómeno aleatorio, y de nuevo vemos aparecer el azar en biología.
Así pues, hay que completar la concepción darwiniana de la evolución por supervivencia de los más aptos señalando también la importancia del azar, que el genetista Motoo Kimura ha resumido con la expresión supervivencia del más afortunado. En la práctica, pues, la evolución es la supervivencia no sólo de los tipos genéticos más aptos, sino también de los que han tenido más suerte.
Parece que la velocidad evolutiva cambia mucho de unos genes a otros, pero no disponemos de mediciones directas. Desconocemos la variación de un gen en el tiempo, porque no sabemos cuál era su frecuencia, pongamos, hace 100 000 años. Pero conocemos bien su variación en el espacio. Siempre hay una conexión bastante estrecha entre la variación temporal y la espacial.
Si conocemos las dimensiones demográficas de las poblaciones humanas a lo largo de toda la historia de la especie, y sus movimientos demográficos a partir del origen del hombre, si sabemos qué genes están sujetos a la selección natural y, si lo están, por qué, entonces podemos predecir la magnitud de su variación geográfica e histórica. Bajo los efectos de la deriva genética, se puede esperar la misma variación media para cualquier gen, pues la causa de la variación (las dimensiones demográficas de la población) es la misma para todos los genes. Pero que un gen varíe más o menos a causa de la deriva, y cuál de ellos lo hace, es aleatorio. Si la selección natural está presente, puede tener un efecto importante, y puede reducir o aumentar de un modo notable la velocidad evolutiva, comparada con la que cabe esperar sólo con la deriva. Pero hay genes que se pueden considerar excluidos de la selección natural. Por ejemplo, podemos prever, conociendo su ADN, que ciertos genes han perdido su funcionalidad. Nos damos cuenta porque mutaciones en su estructura impiden su funcionamiento: por ejemplo, mutaciones de un gen que produce una proteína pueden impedir completamente la producción de esa proteína normal y funcional. Se llaman seudogenes, pero sería mejor llamarlos genes silenciosos. La selección natural no puede influir directamente en ellos, pero, al estar sujetos más directamente a la deriva, nos ayudan a medir su intensidad.
El efecto de la deriva puede estar limitado por otros factores evolutivos. Casi siempre hay intercambios genéticos por migración, a menudo recíproca y mayor entre los vecinos más cercanos, entre pueblos o ciudades vecinos. Esta migración tiende a reducir la variación genética entre pueblos o ciudades. Si fuera lo bastante grande, no habría ninguna diferencia entre los pueblos, las naciones y los continentes. Pero, evidentemente, la migración existente no basta para borrar las diferencias genéticas entre las poblaciones, ya que a menudo son importantes. También es importante la frecuencia de las mutaciones. Si la frecuencia de mutación de un gen es grande, se encuentran muchas formas distintas de él.
Entre los genes con mayor variación espacial están los que producen las inmunoglobulinas (los anticuerpos, que tienen un papel muy importante en la defensa contra las enfermedades infecciosas). Dado que hay diferencias considerables en la distribución geográfica de estas enfermedades, parece normal que los anticuerpos que nos protegen, y por lo tanto los genes que los codifican, difieran de unas regiones a otras. En este caso cabe esperar una situación muy distinta de la producida por la deriva, o sea por el azar, ya que evidentemente la selección natural tiene mucha importancia. Pero la gran variedad de las enfermedades infecciosas, y la de los genes que nos protegen, vuelve a introducir el factor azar por la puerta de atrás. Las enfermedades infecciosas están en continuo cambio a causa de la variación de la virulencia de las bacterias, los virus y los parásitos que las transmiten. De este modo el efecto selectivo puede llegar a ser comparable al de la deriva, en el sentido de que el azar entra en escena a causa de la variación más o menos aleatoria de los agentes de enfermedades infecciosas, sin relación con la acción del tamaño de la población. Se comprende fácilmente, pues, que los resultados del análisis evolutivo obtenidos con las inmunoglobulinas sean cuantitativamente parecidos a los obtenidos cuando se estudia la deriva. Lo mismo se puede decir de otro grupo de genes muy importantes, algunos de los cuales presentan una variación geográfica parecida a la de las inmunoglobulinas: los genes HLA, también implicados en la defensa inmunitaria.
Los genes HLA presentan una gran variedad de formas genéticas. Algunas de ellas tienen poca variabilidad geográfica, y otras mucha. La mayor variación de los HLA se ha observado en muchos grupos indígenas de América del Sur, las poblaciones que muestran la mayor variación en el espacio. En casi todas las demás poblaciones del mundo se encuentra siempre un número elevado de alelos (las distintas formas de los genes HLA), pero en América del Sur el número observado en cada población es menor. Además, en este continente una forma de HLA muy rara en el resto del mundo puede alcanzar una frecuencia alta en una población determinada, mientras que en otra población cercana se encuentra otra forma de HLA con frecuencia inusitadamente elevada. No es fácil descartar la posibilidad de que estas frecuencias altas en algunas poblaciones se deban a condiciones especiales de selección natural (por la presencia local de enfermedades infecciosas que son raras en otras partes). Pero lo más probable, de acuerdo con lo observado en los demás genes, es que se trate de un efecto de la deriva genética.
Por lo tanto, la predicción de la velocidad evolutiva de un gen plantea problemas complejos. De todos modos, el estudio detallado de una población puede ayudar a reconocer si la variación es en buena medida de carácter fortuito, debida a la deriva genética, o resultado de una selección natural variable aleatoriamente, ya que las leyes del azar son bien conocidas y la posibilidad de examinar muchos genes nos ayuda a comprenderlo.
Por el contrario, hay genes que varían muy poco de una población a otra. Es probable que en este caso la selección beneficie al heterocigoto y tienda a estabilizar las frecuencias del polimorfismo y, por lo tanto, a eliminar o reducir su evolución.
A veces una igualdad superficial esconde una gran heterogeneidad. En las zonas de paludismo la talasemia es frecuente, pero hay gran variedad de formas talasémicas, como ha demostrado el análisis molecular. La distribución geográfica de cada una de ellas, el análisis profundo de otros factores genéticos que acompañan a la talasemia, y los conocimientos históricos sobre las regiones donde están difundidas, permiten asociar algunos de estos tipos genéticos particulares a migraciones conocidas, como la fenicia y griega en el Mediterráneo y las polinesias en el Pacífico.
La mayor parte de los genes tienen una variación geográfica intermedia entre la más elevada de los genes relacionados con la inmunidad contra las enfermedades infecciosas y la reducida, que es característica de algunos genes con la ventaja de los heterocigotos. Es probable que los polimorfismos de variación geográfica intermedia sean selectivamente neutros o casi neutros, y que por tanto su evolución se deba en gran medida a la deriva genética. En efecto, su variación tiene un nivel parecido al que cabría esperar a partir de nuestros escasos conocimientos de la demografía del hombre moderno en los últimos 100 000 años.
Nuestra labor se vería muy facilitada si pudiéramos estar seguros de que la velocidad evolutiva, calculada a partir del promedio de muchos genes, es la misma en todas las ramas del árbol evolutivo. Hemos dado alguna idea sobre los factores que pueden influir en la velocidad evolutiva. ¿Hay algún modo de comprobar si la situación es tan sencilla como en esta hipótesis?
En este capítulo hemos visto un cuadro que ilustra las distancias genéticas entre los continentes. En él vemos que África es el continente más alejado genéticamente de los demás. La distancia media entre África y los otros cuatro continentes es casi el doble que la que separa Oceanía de los otros tres (21,1 ± 1,7 frente a 12,7 ± 1,4; las cantidades después del ± son el error estándar de las dos medias, y en este caso nos dicen que la diferencia entre ellas va mucho más allá del error estadístico). Las otras distancias son inferiores. Más adelante veremos que hay una excelente razón histórica que puede explicar este hecho. Para examinar el problema de la constancia de la velocidad evolutiva, veamos las distancias entre África y los demás continentes, que son las mayores:
24,7 con Oceanía
20,6 con Asia
16,6 con Europa
22,6 con América
La distancia menor es la que separa África de Europa, seguida de la que hay entre África y Asia. Si la velocidad evolutiva fuera realmente constante, cabría esperar que los cuatro valores fueran los mismos, salvo naturalmente la variación estadística[14].
La distancia con Europa es anormalmente pequeña. La explicación más sencilla es que entre los dos continentes, que están muy próximos, se produjeron intercambios genéticos importantes, es decir, migraciones en ambas direcciones. También Asia, la otra vecina de África, está más cerca de ella genéticamente que América y Oceanía, aunque menos que Europa. Se observa el mismo fenómeno al comparar las distancias genéticas entre Oceanía y los otros tres continentes: de nuevo, la distancia menor es entre los dos continentes más próximos geográficamente, Oceanía y Asia. También sucede esto con la última comparación posible, la de Asia y América, los dos continentes geográfica y genéticamente más cercanos (y también históricamente).
Vemos, pues, que la expectativa de una velocidad de evolución constante no se cumple a rajatabla, pero la diferencia no es grande. La causa debe de ser el intercambio genético entre poblaciones, que altera las distancias genéticas de algunas de ellas, reduciendo las que hay entre poblaciones que han tenido intercambios migratorios importantes. La migración está casi siempre limitada a distancias geográficas bastante pequeñas, pero en períodos largos también puede quedar reflejada en distancias mayores, y el problema es interesante y complicado, por lo que merece una digresión.
El hombre se pasa la vida moviéndose y desplazándose. Durante la mayor parte de su historia fue cazador y recolector; luego, en los últimos 10 000 años, se hizo agricultor y ganadero. Los territorios de caza no solían estar muy alejados entre sí, y probablemente no cambiaban muy a menudo. Entre los pigmeos africanos, los territorios de caza son la heredad del grupo (la «banda de caza»), y cada nutrido tiene derecho a incorporarle el de su mujer. Hay, pues, buenas razones para «desplazarse lejos», una regla pigmea que, además de tener la ventaja económica de ampliar la propia esfera de influencia, disminuye la probabilidad de un matrimonio con un pariente demasiado estrecho. Como casi todos los pueblos con economía primitiva, los pigmeos procuran no casarse entre primos hermanos, pero no tienen en cuenta los vínculos de parentesco más lejanos. Las actividades de caza y recolección requieren una movilidad algo superior que la de los campesinos; pero la diferencia no es grande, por lo menos a juzgar por los datos obtenidos actualmente en África. Por su parte, los ganaderos a veces recorren, entre el verano y el invierno, distancias grandes, de 500 ó 1000 km, pero para estas migraciones estacionales casi siempre utilizan los mismos pastos, año tras año (la trashumancia). Casi nunca se trata de un nomadismo que implique un «paseo al azar». Estos cambios de residencia por razones de trabajo continúan hoy, pero no se suelen hacer en grupo, sino individualmente. De todos modos hay, y ha habido, probablemente durante decenas de miles de años, otras causas de desplazamiento: para participar en ferias, fiestas, etc. Una razón de cambio de residencia permanente es el matrimonio, ya que por lo menos uno de los esposos, por lo general la mujer, tiene que desplazarse para unirse al otro. Hay otras causas, como el trabajo, pero excepto en el último siglo, con su proliferación de medios de transporte, los movimientos han sido limitados. Entre los pigmeos los desplazamientos de un campamento a otro requieren uno o varios días de viaje. Un día de recorrido a pie significa 30 ó 40 kilómetros, y 20 si van cargados.
Desde el punto de vista genético, los desplazamientos importantes son los cambios de residencia permanentes, sobre todo los de una familia entera, y más aún el desplazamiento que acompaña al matrimonio. Los mejores datos que tenemos sobre la distancia media entre los lugares de nacimiento del marido y la mujer indican:
1. 30-50 km para los cazadores-recolectores de las zonas tropicales (probablemente mucho mayores pero desconocidas para aquellos que, como los esquimales, ocupan regiones árticas con densidad de población muy baja);
2. 10-20 km para los agricultores de las regiones africanas con baja densidad de población;
3. 5-10 km para los agricultores europeos de los últimos siglos.
Debemos añadir que hoy la distancia media entre los lugares de nacimiento del marido y la mujer aumenta con velocidad creciente desde mediados de siglo, gracias a la extensión de los ferrocarriles y a la aparición de los otros medios modernos de transporte.
Esta «pequeña» migración de carácter individual o familiar es muy frecuente, pero suele limitarse a distancias pequeñas. Es la principal causa de la relación entre distancia geográfica y distancia genética, que (como hemos visto en el capítulo anterior) presenta importantes regularidades, comunes a todos los continentes, pero también diferencias cuantitativas entre distintas regiones. Pero la migración pequeña no nos ayuda a explicar las diferencias entre continentes, que deben su existencia a la acumulación de muchas «pequeñas» migraciones, o bien a «grandes» migraciones, responsables de colonizaciones de nuevos territorios, a veces a gran distancia.
Las grandes migraciones son un fenómeno completamente distinto de las pequeñas. Son sucesos poco frecuentes, que han tenido mucha influencia en la historia de la especie. Son migraciones a menudo repetidas de grupos de individuos, pequeños o grandes, que se establecen en un nuevo lugar, a veces muy alejado del lugar de origen. Podemos llamarlas colonizaciones. Conocemos muchos ejemplos históricos: la colonización griega y fenicia en el Mediterráneo, la blanca en América, Australia y Suráfrica, o la colonización china del sureste asiático. En época casi histórica, la malaya-polinesia en el Pacífico y el Índico. Antes del comienzo de la historia seguramente hubo muchos casos parecidos, y revelaremos alguno que no era conocido antes de nuestras investigaciones genéticas.
En época histórica las colonizaciones están organizadas, y lo que las desencadena es la superpoblación de la región de origen. A veces se trataba de grupos sociales que emigraban para huir de persecuciones, por ejemplo de las religiosas. Las hambres y las guerras han sido otras causas de sucesos de este tipo. Cuando hay un aumento demográfico, puede producirse una supersaturación de la colonia, que inevitablemente conduce a más migraciones. Por ejemplo, el norte de América fue ocupado a principios del siglo XVIII sólo en la costa oriental, y la expansión hacia la costa occidental duró más de dos siglos. La repetición del ciclo crecimiento-migración a las nuevas regiones ocupadas puede llevar a grandes expansiones, que pueden durar siglos o milenios y ocupar territorios muy extensos.
En el capítulo 4 veremos que estas expansiones han formado estructuras muy características en el mapa geográfico de los genes. El enfoque geográfico del estudio de la variación genética es muy distinto del enfoque con los árboles evolutivos, y ha creado problemas nuevos. En el estudio de los árboles elegimos un pequeño número de poblaciones y nos planteamos la cuestión de sus orígenes históricos. Dado que todos los hombres del planeta tienen un origen común, podemos esperar que todos desciendan de una misma población, cuyo número creció mucho y empezó a fragmentarse a partir del punto de origen. Esta difusión hizo que la población se extendiera por otros continentes y, al final, por todo el mundo. El paso de un área geográfica a otras bastante aisladas de la primera puede implicar discontinuidades en el proceso de difusión, fisiones que pueden ser análogos físicos de las horcaduras de un árbol.
La naturaleza de un proceso de difusión sugiere que las poblaciones vecinas probablemente estén destinadas a tener muchos intercambios genéticos, más o menos recíprocos, debidos a pequeñas o grandes migraciones. Estas mezclas entre ramas pueden hacer que el modelo de un árbol con bifurcaciones sucesivas sea demasiado simple para representar el proceso evolutivo del hombre. En efecto, algunos métodos de reconstrucción de los árboles se prestan menos que otros a esta representación, al estar muy sujetos al efecto de las mezclas. De todos modos, resultan útiles para identificarlas.
Un control importante de la validez de la reconstrucción del árbol evolutivo es si todos los genes o caracteres que se pueden emplear para este mismo fin llevan al mismo resultado. Si hay diferencias, se tienen que poder explicar de una forma razonable. En general, las pruebas estadísticas de la estabilidad de los resultados, por ejemplo con distintas clases de polimorfismos, como grupos sanguíneos, enzimas, ADN, etc., han revelado que los árboles reconstruidos son siempre más o menos los mismos. Hay algunas ramas que no se repiten con tanta claridad, a causa de la insuficiencia de poblaciones y genes examinados. Pero unos grupos de genes completamente distintos, si son lo bastante numerosos, dan árboles muy parecidos o idénticos. Los polimorfismos genéticos más conocidos (los que llamamos clásicos) se estudian en las proteínas o, en general, en los productos de los genes, y no directamente en éstos. Los polimorfismos más modernos, utilizados después de 1981-1982, permiten el estudio directo de los genes (es decir, del ADN). Presentan muchas ventajas con respecto a los clásicos, y una sola desventaja, que hasta ahora se han estudiado en pocas poblaciones, mientras que de los más antiguos hay datos sobre cientos y miles de poblaciones distintas.
Aún quedan problemas prácticos por resolver, para reconstruir la evolución del hombre a través del estudio de los seres vivos, y también de los fósiles. El próximo capítulo está dedicado al análisis de cuestiones relacionadas con la comparación de datos obtenidos con polimorfismos, así como de los resultados de estudios arqueológicos, que pueden ayudarnos a reconstruir el pasado con otros medios.